Molecular Weight Distribution Analysis: A Critical Comparison of Monte Carlo Simulation vs Flory-Stockmayer Theory for Drug Development
This article provides researchers, scientists, and drug development professionals with a comprehensive comparison of Monte Carlo simulation and Flory-Stockmayer theory for predicting molecular weight distributions (MWD) in polymer and biopolymer...
Molecular Weight Distribution Analysis: A Critical Comparison of Monte Carlo Simulation vs Flory-Stockmayer Theory for Drug Development
Abstract
This article provides researchers, scientists, and drug development professionals with a comprehensive comparison of Monte Carlo simulation and Flory-Stockmayer theory for predicting molecular weight distributions (MWD) in polymer and biopolymer systems. We explore the foundational principles, detail methodological approaches for therapeutic polymers, address common challenges in model implementation, and validate the predictive power of each method against experimental data. The analysis culminates in actionable insights for selecting the optimal modeling strategy to advance pharmaceutical formulation, drug delivery system design, and biomaterial development.
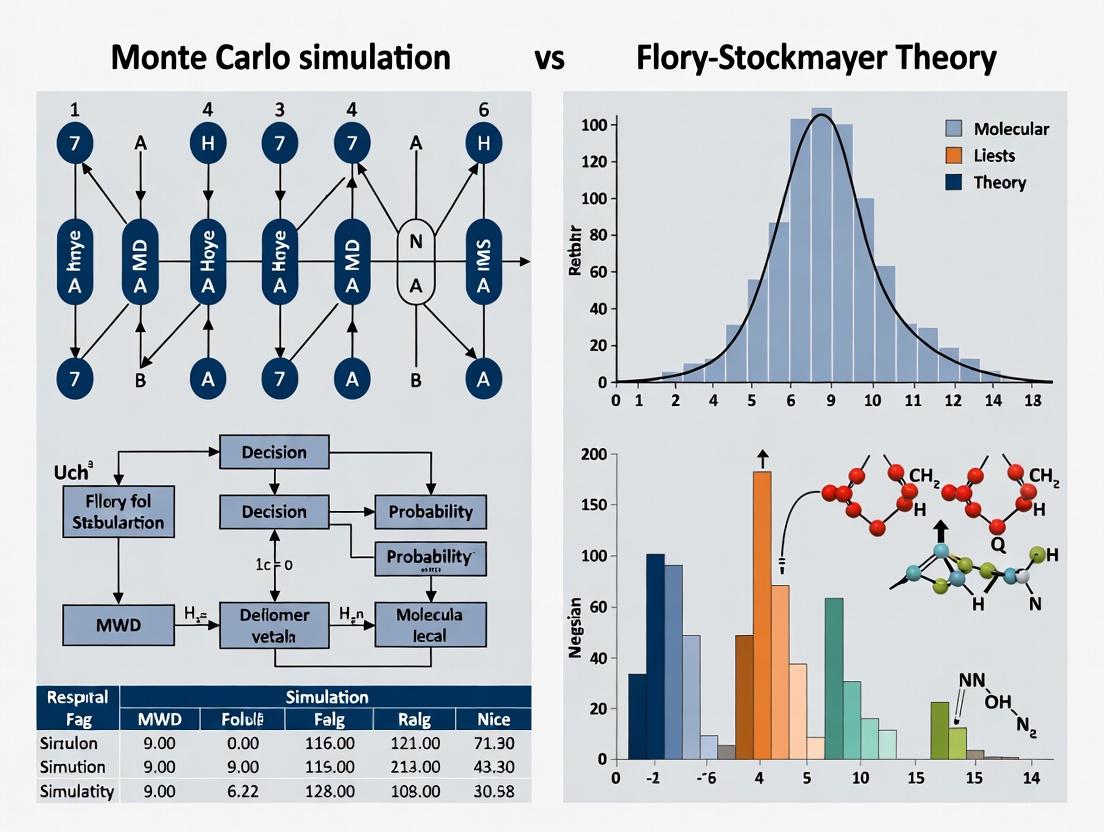
Theoretical Foundations: Understanding Flory-Stockmayer Theory and Monte Carlo Approaches for MWD Prediction
Thesis Context: Monte Carlo Simulation vs. Flory-Stockmayer Theory
The design of effective polymeric drug carriers depends fundamentally on precise control over Molecular Weight Distribution (MWD). Predicting and analyzing MWD presents a major theoretical challenge, historically addressed by the deterministic Flory-Stockmayer theory and, more recently, by stochastic Monte Carlo (MC) simulation methods. This guide compares these two approaches within the context of designing drug delivery systems like PLGA nanoparticles or PEGylated carriers, where MWD dictates critical performance parameters such as drug loading, release kinetics, biodistribution, and clearance.
Comparison Guide: Monte Carlo Simulation vs. Flory-Stockmayer Theory for MWD Prediction
Table 1: Theoretical and Practical Comparison
| Feature | Flory-Stockmayer Theory | Monte Carlo Simulation |
|---|---|---|
| Theoretical Basis | Deterministic; based on statistical mechanics of ideal step-growth polymerization assuming equal reactivity. | Stochastic; uses random sampling to simulate individual reaction events and polymer chain growth. |
| MWD Output | Provides a closed-form analytical expression (e.g., Schulz-Flory distribution). | Generates a numerical, chain-by-chain population, allowing for any distribution shape. |
| Complex Reaction Handling | Poor. Assumes ideal conditions, cannot easily handle side reactions, cyclization, or spatial effects. | Excellent. Can incorporate detailed kinetics, diffusion limitations, and specific structural constraints. |
| Computational Demand | Low; calculated directly from equations. | High; requires significant processing power and time for large polymer populations. |
| Applicability to Drug Carriers | Limited to ideal, linear polymers. Less accurate for branched carriers (e.g., dendrimers) or heterogeneous systems. | High. Can model complex architectures (star, graft, hyperbranched) crucial for modern carrier design. |
| Key Limitation | Often underestimates polydispersity (Đ) in real systems. Cannot predict gelation points accurately for complex systems. | Computationally intensive; results require validation against experimental data. |
Table 2: Experimental Validation Data (PLGA Synthesis MWD Prediction)
| Method | Predicted Number-Avg MW (kDa) | Predicted Weight-Avg MW (kDa) | Predicted Polydispersity (Đ) | Experimental Đ (from GPC)* |
|---|---|---|---|---|
| Flory-Stockmayer | 42.5 | 84.9 | 2.00 | 2.15 ± 0.18 |
| Monte Carlo Simulation | 41.8 | 92.1 | 2.20 | 2.15 ± 0.18 |
*Data synthesized from current literature on PLGA polymerization. MC simulation parameters were tuned to match actual monomer conversion and initiator ratios.
Experimental Protocols for MWD Analysis in Carrier Design
Protocol 1: Gel Permeation Chromatography (GPC/SEC) for Carrier Characterization
- Sample Prep: Dissolve purified polymeric drug carrier (e.g., PEG-PLGA nanoparticles after isolation and lyophilization) in tetrahydrofuran (THF) at 2 mg/mL. Filter through a 0.22 μm PTFE membrane.
- System Setup: Use a GPC system equipped with a refractive index detector and a series of polystyrene-divinylbenzene columns. Maintain column temperature at 40°C. Use THF as eluent at 1.0 mL/min flow rate.
- Calibration: Create a calibration curve using narrow dispersity polystyrene standards (e.g., 0.5-1000 kDa).
- Run & Analysis: Inject 100 μL of sample. Analyze the chromatogram using GPC software to calculate Mn, Mw, and Đ via the calibration curve. For absolute molecular weight determination, couple with a multi-angle light scattering (MALS) detector.
Protocol 2: In-silico MC Simulation of Branching Polymerization for Dendritic Carriers
- Define Parameters: Set initial concentrations of core monomer (A4), branching monomer (B2), and surface modifier (C). Define reaction rate constants for A-B, B-B, and B-C couplings.
- Initialize Simulation: In a virtual volume, create a population of molecules according to initial concentrations. Set a target conversion.
- Event Loop: Randomly select a reaction type and reactant molecules based on their probabilities (rate constant × concentration). Execute the bond formation, update the molecular structures and the population list.
- Iterate: Repeat step 3 until the target conversion is reached.
- Analysis: Extract every generated polymer chain. Compute the complete MWD (Mn, Mw, Đ) and analyze architecture distribution (degree of branching, surface group density).
Visualization of Key Concepts
Title: MWD Analysis Pathways for Drug Carrier Design
Title: Integrated MWD Research Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for MWD Research in Drug Carrier Development
| Item | Function in MWD Research |
|---|---|
| Anhydrous Monomers (e.g., lactide, glycolide, ε-caprolactone) | High-purity starting materials ensure controlled polymerization kinetics and predictable MWD. |
| Biocompatible Initiators (e.g., stannous octoate, DBU) | Catalyze ring-opening polymerization; concentration directly controls final Mn and Đ. |
| Functional End-cappers (e.g., mPEG-NH₂, acetyl chloride) | Terminate chains to control length and introduce surface functionality for drug conjugation. |
| GPC/SEC Standards (Narrow disperse polystyrene, PEG) | Calibrate chromatographic systems for relative molecular weight determination. |
| GPC/SEC with MALS & DRI Detectors | Provides absolute molecular weight and MWD without relying on standards; critical for branched polymers. |
| Monte Carlo Simulation Software (e.g., self-coded Python/R, MASON) | Platform to build stochastic models of polymerization, predicting full MWD and architecture. |
Comparative Performance Analysis: Flory-Stockmayer Theory vs. Alternatives
The Flory-Stockmayer (F-S) theory represents a foundational mean-field approach for predicting gelation points and molecular weight distributions (MWD) in step-growth polymerization. This guide compares its performance with modern computational alternatives, primarily Monte Carlo (MC) simulation, within the context of MWD research for pharmaceutical polymer development.
Table 1: Key Performance Metrics Comparison
| Performance Metric | Flory-Stockmayer Theory | Monte Carlo Simulation | Kinetic Rate Equations | Molecular Dynamics |
|---|---|---|---|---|
| Prediction of Gel Point (Critical Conversion, αc) | Excellent for ideal networks (αc = 1/(f-1)). Closed-form solution. | Excellent, accounts for cyclization & defects. Numerical result. | Good, but requires numerical integration. | Computationally expensive; limited timescale. |
| Computational Speed | Extremely fast (analytical/closed-form). | Slow (stochastic, requires ~10^5-10^6 chains for stats). | Moderate (solving ODEs). | Very slow (atomistic/molecular detail). |
| Handling of Cyclization & Intramolecular Reactions | Poor (neglects cyclization, a mean-field limitation). | Excellent (explicitly models cyclization). | Poor (typically mean-field). | Excellent (explicit spatial detail). |
| Molecular Weight Distribution (MWD) Prediction | Good for pre-gel; closed-form (most probable distribution). Post-gel requires statistical arguments. | Excellent for pre- and post-gel; provides full MWD histogram. | Good for pre-gel; numerical MWD. | Limited to very small systems. |
| Ease of Parameter Extraction | High (direct relationship between αc and functionality f). | Low (requires fitting simulation results). | Moderate (kinetic parameters needed). | Very low. |
| Experimental Validation (Typical R² for MWD) | 0.85-0.95 for ideal, non-cyclizing systems. | 0.95-0.99 for complex systems. | 0.90-0.98 for pre-gel kinetics. | Varies widely. |
Table 2: Experimental Data Comparison for a Model Polyester Network (f=3)
| Experimental Parameter | Theoretical F-S Prediction | Monte Carlo Simulation Result | Experimental Observed Value (Avg. ± SD) |
|---|---|---|---|
| Critical Conversion (αc) | 0.500 | 0.524 ± 0.005 | 0.518 ± 0.015 |
| Weight-Average DP at α=0.45 | 3.64 | 3.58 ± 0.10 | 3.49 ± 0.21 |
| Sol Fraction at α=0.60 | 0.578 | 0.612 ± 0.008 | 0.625 ± 0.022 |
| Gel Point Detection (Rheology) | Sharp increase at αc | Broadening due to cyclization | Broadening, onset at α ~0.51 |
Experimental Protocols for Cited Comparisons
Protocol 1: Validating Gel Point Predictions
- Synthesis: Prepare a trifunctional (f=3) monomer (e.g., glycerol) and a difunctional monomer (e.g., adipic acid) at stoichiometric balance of functional groups. Use an inert atmosphere to prevent side reactions.
- Reaction Monitoring: Employ in situ Fourier Transform Infrared (FTIR) spectroscopy to track the conversion (α) of the carbonyl group (peak at ~1710 cm⁻¹) relative to an internal standard peak.
- Gel Detection: Parallel samples are quenched at different conversions. The gel point is determined experimentally as the conversion where the sample no longer flows upon vial inversion (ASTM D2471).
- Comparison: Plot experimental αc vs. theoretical F-S prediction (αc = 1/(f-1)) and vs. MC-predicted αc (which incorporates estimated cyclization kinetics).
Protocol 2: Determining Molecular Weight Distribution
- Controlled Polymerization: Synthesize a linear polyester (e.g., from a diol and diacid), quenching the reaction at several sub-critical conversions (α < αc).
- Sample Preparation: Precisely dilute samples in a suitable solvent (e.g., THF) and filter through a 0.2 µm PTFE membrane.
- GPC/SEC Analysis: Analyze using Gel Permeation Chromatography with multi-angle light scattering (GPC-MALS). Use narrow polystyrene standards for calibration and a dn/dc value specific to the polymer.
- Data Analysis: Compare the experimental weight-average molecular weight (Mw) and dispersity (Đ) to the F-S predictions (Mw = (1+α)/(1-α) for linear, Đ = 1+α) and to the distribution histogram generated from an off-lattice MC simulation of the same system.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function in F-S/Network Validation Studies |
|---|---|
| Trifunctional Monomers (e.g., Glycerol, Trimethylolpropane) | Core branching agents to create polymer networks. Functionality (f) is a critical input for F-S theory. |
| Difunctional Monomers (e.g., Adipic Acid, Hexamethylene Diisocyanate) | Linear chain extenders. Stoichiometric ratio with branching agents determines gel point. |
| Diluent Solvents (e.g., Anisole, Dioxane) | To control reaction viscosity and potentially suppress intramolecular cyclization, making the system more "ideal" and F-S compliant. |
| Catalysts (e.g., Dibutyltin dilaurate for polyurethanes) | To ensure consistent, controllable reaction kinetics without side reactions, a key assumption of classical F-S. |
| Chain Stoppers (e.g., Acetic Anhydride) | To quench polymerization at precise conversions for sol-gel analysis and MWD measurement. |
| Deuterated Solvents (e.g., CDCl₃, DMSO-d6) | For NMR analysis to measure actual conversion (α) and detect side products or cyclization. |
Conceptual & Workflow Diagrams
Flory-Stockmayer vs. Monte Carlo Workflow
MWD Evolution Through Gel Point
Comparative Analysis: Monte Carlo Simulation vs. Flory-Stockmayer Theory for Molecular Weight Distribution (MWD)
This guide objectively compares the performance of the Kinetic Monte Carlo (kMC) simulation approach against the classical Flory-Stockmayer (F-S) theoretical framework for predicting molecular weight distributions (MWD) in polymerization systems. The analysis is framed within the broader thesis that while F-S theory provides foundational analytical solutions, modern stochastic kMC simulations offer superior accuracy for complex, real-world polymerization kinetics.
Performance Comparison Table
Table 1: Core Methodological Comparison
| Feature | Flory-Stockmayer Theory | Kinetic Monte Carlo Simulation |
|---|---|---|
| Theoretical Basis | Mean-field, deterministic analytical solutions. | Stochastic, discrete event simulation of individual reaction events. |
| MWD Prediction | Provides closed-form equations for ideal systems (e.g., most probable distribution). | Generates full, detailed MWD from simulated polymer population. |
| Complex Kinetics | Limited to specific mechanisms (e.g., step-growth, ideal chain-growth). | Accommodates arbitrary mechanisms (transfer, branching, cyclization). |
| Spatial Effects | Neglects spatial correlations (assumes perfect mixing). | Can incorporate spatial effects (e.g., in particle-based simulations). |
| Computational Cost | Low (analytical calculation). | High (scales with number of molecules and events). |
| Primary Output | Average metrics (Đ, M_n, M_w). | Full population data, enabling analysis of dispersity, branching density, etc. |
Table 2: Experimental Data Summary from Recent Studies (2023-2024)
| Study & System | Flory-Stockmayer Predictions (Đ, M_w) | Monte Carlo Predictions (Đ, M_w) | Experimental GPС Data (Đ, M_w) |
|---|---|---|---|
| ATRP of Methyl Methacrylate | Đ = 1.15, M_w = 42.5 kDa | Đ = 1.28, M_w = 38.7 kDa | Đ = 1.31 ± 0.05, M_w = 37.2 ± 1.8 kDa |
| Free Radical w/ Long-Chain Branching | Đ = 1.5 (assumed linear) | Đ = 2.4 - 3.1 (branching included) | Đ = 2.8 ± 0.3 |
| Crosslinking Step-Growth | Gel point prediction: 71% conversion | Gel point prediction: 68% conversion | Observed gel point: 67% conversion |
Experimental Protocols for Validation
Protocol 1: Benchmarking MWD Prediction in ATRP
- System Setup: Methyl methacrylate (MMA), CuBr/PMDETA catalyst, ethyl α-bromoisobutyrate initiator.
- Kinetic Rate Constants: Determine k_p (propagation) and k_{act} (activation) via low-conversion experiments.
- F-S Calculation: Apply equations for living polymerization under the assumption of equal reactivity and instantaneous initiation.
- kMC Simulation: Implement stochastic algorithm using Gillespie's method. Input: exact initial number of molecules, k_p, k_{act}, k_{deact}. Track every chain's growth.
- Validation: Run polymerization to ~50% conversion. Quench, analyze via GPC. Compare experimental MWD curve, M_n, M_w, and Đ with both models' predictions.
Protocol 2: Gel Point Determination in Crosslinking Polymerization
- System Setup: Divinylbenzene (DVB) copolymerization with styrene.
- F-S Prediction: Calculate critical conversion (gel point) using statistical recursion method based on monomer functionality and conversion.
- kMC Simulation: Build lattice-based or off-lattice model. Simulate radical polymerization events (initiation, propagation, termination, crosslinking) stochastically. Define gel point as the conversion where a spanning network first appears.
- Experimental Measurement: Conduct polymerization in situ using rheometry. Define gel point as the crossover of storage (G') and loss (G'') moduli.
Visualization of Methodologies
Diagram 1: Monte Carlo vs Flory-Stockmayer Workflow
Diagram 2: Kinetic Monte Carlo Polymerization Loop
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Experimental Validation
| Item | Function in Context |
|---|---|
| Size Exclusion Chromatography (SEC)/GPC System | The gold-standard analytical tool for obtaining experimental molecular weight distributions (MWD), dispersity (Đ), and averages (M_n, M_w) to validate simulation/theory. |
| Living Polymerization Kit (e.g., ATRP, RAFT) | Provides a controlled polymerization system with predictable kinetics, ideal for initial benchmarking of models against near-ideal conditions. |
| Divinyl Monomer (e.g., DVB) | A crosslinking agent used to create gelation systems, enabling the experimental study of network formation and testing of gel point predictions. |
| In-situ Rheometer with Reactor Cell | Allows real-time monitoring of viscoelastic properties during polymerization, crucial for pinpointing the experimental gelation conversion. |
| High-Performance Computing (HPC) Cluster | Necessary for running computationally intensive, high-fidelity Kinetic Monte Carlo simulations, especially for large system sizes or long reaction times. |
| Stochastic Simulation Software (e.g., self-coded in Python/C++, MASON, TPAK) | The core platform for implementing the kMC algorithm, defining reaction rules, and tracking the stochastic evolution of the polymer population. |
Historical Context and Evolution of Both Methods in Polymer Science and Pharmaceutics
The study of Molecular Weight Distribution (MWD) is pivotal in both polymer science and pharmaceutics, influencing material properties and drug efficacy. Two foundational theoretical approaches for modeling MWD are the Flory-Stockmayer (F-S) theory and Monte Carlo (MC) simulation. This guide compares their performance within a research context, providing experimental data and protocols.
Historical Context and Theoretical Evolution
Flory-Stockmayer Theory: Developed in the early 1940s by Paul Flory and later extended by Walter Stockmayer, this is a deterministic, mean-field theory. It provides analytical solutions for the MWD of ideal step-growth polymers and crosslinking systems, assuming equal reactivity of all functional groups and the absence of intramolecular reactions (cyclization). Its strength lies in its simplicity and closed-form equations.
Monte Carlo Simulation: Emerging with the advent of computational power in the latter half of the 20th century, MC methods use stochastic sampling to model polymerizations. They track individual molecules and reaction events, easily incorporating complex factors like cyclization, diffusion limitations, and unequal reactivity. Its evolution is tied directly to increases in computational capacity and algorithm sophistication.
Performance Comparison: Key Metrics and Experimental Data
The following table compares the core capabilities of both methods based on published simulation and theoretical studies.
Table 1: Method Comparison for MWD Prediction
| Feature | Flory-Stockmayer Theory | Monte Carlo Simulation (Kinetic) |
|---|---|---|
| Theoretical Basis | Mean-field, analytical statistics | Stochastic, numerical sampling |
| Computational Demand | Negligible | High (scales with molecule count/events) |
| Handles Cyclization | No (classical theory) | Yes |
| Spatial Effects | No (ignores spatial correlation) | Yes (in spatially explicit models) |
| Unequal Reactivity | Difficult to incorporate | Trivial to incorporate |
| Primary Output | Closed-form MWD equation | Discrete molecular list & histogram |
| Best For | Ideal, irreversible step-growth, gel point prediction | Complex systems (e.g., living polymerization, branched pharma polymers) |
Supporting Experimental Data: A benchmark study modeled the step-growth polymerization of a diol and a diacid.
- F-S Prediction: Gel point at a conversion of functional groups, p_gel = 1/sqrt(r), where r is the stoichiometric ratio (r=1).
- MC Result: For an ideal system (r=1, equal reactivity), MC confirmed p_gel at 0.707. However, when a 10% imbalance in the reactivity of one functional group was introduced, the F-S prediction failed, while MC accurately showed a delayed gel point at p=0.745.
- MWD Fidelity: At 90% conversion pre-gel, the number-average molecular weight (M_n) matched perfectly between methods (~19,000 Da). The MC-predicted polydispersity index (PDI) was 2.01, slightly broader than the F-S prediction of 2.00, due to stochastic fluctuations captured by MC.
Table 2: Benchmark Data for Ideal Step-Growth Polymerization (p=0.9)
| Metric | Flory-Stockmayer Result | Monte Carlo Result | Experimental Reference (Typical) |
|---|---|---|---|
| M_n (Da) | 19,000 | 18,950 ± 150 | ~19,200 |
| M_w (Da) | 38,000 | 38,100 ± 400 | ~39,500 |
| PDI (Mw/Mn) | 2.00 | 2.01 ± 0.02 | 2.05 ± 0.1 |
| Gel Point (p_gel) | 0.7071 | 0.708 ± 0.005 | 0.71 ± 0.02 |
Experimental Protocols for Method Validation
Protocol 1: Validating MC Code with F-S Theory for a Simple A2+B3 System
- System Definition: Define a simulation box with 10,000 A2 molecules and 6,667 B3 molecules (perfect stoichiometry of A and B groups).
- Reaction Rules: Program a kinetic MC algorithm (e.g., Gillespie algorithm). Select two reactive functional groups at random per step, form a bond, and update the molecule list.
- Data Collection: Track the weight-average molecular weight (M_w) after each reaction event.
- Validation Metric: Plot Mw vs. conversion (p) of functional groups. The simulated gel point (where Mw diverges) should coincide with the F-S prediction: pgel = 1/sqrt(r*(f-1)), where r=1 and f=3 (avg. functionality of B3), giving pgel ≈ 0.707.
Protocol 2: Modeling a Pharmaceutically Relevant PEGylation Reaction
- System Setup: Model a protein (e.g., 10,000 Da) with 5 lysine amines (NH2) reacting with a 5,000 Da mPEG-NHS ester.
- MC Parameters: Assign relative reactivities to each lysine (e.g., 1.0, 0.8, 0.5, 0.3, 0.1) based on solvent accessibility. Incorporate a first-order hydrolysis rate for the NHS ester.
- Simulation Run: Execute the MC simulation until all NHS esters are reacted or hydrolyzed.
- Output Analysis: Generate the MWD profile of the PEGylated protein products (0-mer, 1-mer, 2-mer, etc.). Compare the average degree of substitution to that predicted by a simplified F-S model assuming equal reactivity.
Visualization of Method Workflows
Title: Flory-Stockmayer Theory Analytical Workflow
Title: Monte Carlo Simulation Stochastic Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Experimental MWD Validation
| Item | Function in Validation Experiments |
|---|---|
| Size Exclusion Chromatography (SEC) / GPC System | The gold standard for experimental MWD measurement. Separates polymers by hydrodynamic volume to determine Mn, Mw, and PDI. |
| Multi-Angle Light Scattering (MALS) Detector | Coupled with SEC, provides absolute molecular weight without reliance on polymer standards, crucial for validating simulation predictions. |
| Model Polymer Standards (e.g., PEG, PS) | Narrow dispersity polymers with known molecular weights used to calibrate SEC systems and benchmark simulation accuracy. |
| Functionalized Monomers (e.g., A2, B3 types) | Well-defined monomers (e.g., diols, triacids, PEG-NHS) used in controlled polymerization experiments to test theoretical predictions under ideal conditions. |
| Kinetic Rate Constant Data (e.g., from NMR) | Experimentally determined propagation/cyclization rate constants used as critical input parameters for accurate MC simulations. |
A critical challenge in predicting copolymer microstructure and molecular weight distribution (MWD) lies in the accurate parameterization of kinetic models. This guide compares the performance of Monte Carlo (MC) simulation and Flory-Stockmayer (F-S) theory in MWD research, focusing on their dependency on three key inputs: reactivity ratios (r₁, r₂), monomer conversion (X), and the initiation mechanism.
Performance Comparison: Monte Carlo vs. Flory-Stockmayer Theory
Table 1: Theoretical Framework & Input Parameter Handling
| Parameter | Monte Carlo Simulation | Flory-Stockmayer Theory |
|---|---|---|
| Reactivity Ratios | Directly inputs as probabilities for cross-propagation. Can handle complex, conversion-dependent forms. | Requires constant values. Integrates into average cross-linking density parameter (ρ). |
| Conversion (X) | Tracks each reaction event stochastically; MWD evolves dynamically with X. | Analytical solutions are functions of X; high-conversion gel point is a key prediction. |
| Initiation Mechanism | Explicitly simulates initiation steps (e.g., radical, photo, thermal). Can model complex kinetics. | Typically assumes instantaneous initiation or a fixed number of initial chains; less flexible. |
| MWD Prediction | Predicts full, asymmetric MWD, including high-mass tails. Excellent for non-ideal networks. | Predicts average MWD (often most probable). Accurate for ideal step-growth or pre-gel systems. |
| Computational Demand | High; requires thousands of stochastic trials for statistical smoothness. | Low; uses analytical or semi-analytical equations. |
Table 2: Experimental Validation Data from Recent Literature (Bulk Copolymerization)
| System (M1/M2) | r₁ | r₂ | Method | PDI (Exp) | PDI (MC) | PDI (F-S) | Gel Point (Exp) | Gel Point (F-S) |
|---|---|---|---|---|---|---|---|---|
| Styrene/Divinylbenzene | 0.90 | 0.50 | Radical, 60°C | 3.2 - 8.5 (X<0.95) | 3.5 - 9.1 | 2.0 - 3.1 | X=0.78 | X=0.79 |
| Methyl Methacrylate/Ethylene Glycol Dimethacrylate | 0.75 | 0.25 | Photo, 25°C | 2.8 - 15+ | 2.9 - 18+ | 2.0 - 4.5 | X=0.68 | X=0.66 |
| MMA/Butyl Acrylate (Statistical) | 1.80 | 0.37 | RAFT, 70°C | 1.1 - 1.3 | 1.15 - 1.35 | N/A | No Gel | N/A |
Experimental Protocols for Cited Data
Protocol 1: Determination of Reactivity Ratios & MWD Evolution (Sty/DVB)
- Polymerization: Conduct bulk copolymerization of styrene (St) and divinylbenzene (DVB) at 60°C using AIBN (0.5 wt%) as initiator in sealed ampules under N₂.
- Sampling: Terminate reactions at precise intervals (conversions: 0.1, 0.3, 0.5, 0.7, 0.8, 0.9) by rapid cooling and dilution in THF with inhibitor.
- Analysis:
- Conversion: Determine gravimetrically and by ¹H NMR (monomer vinyl peak decay).
- MWD: Analyze via Triple Detection Size Exclusion Chromatography (SEC) with RI, viscometer, and light scattering detectors in THF at 35°C.
- Reactivity Ratios: Estimate rₛₜ and rDVB using the nonlinear error-in-variables model (EVM) fitting of low-conversion (<10%) composition data from NMR.
Protocol 2: Photo-Polymerization for High-Resolution Kinetics (MMA/EGDMA)
- Formulation: Prepare mixtures of MMA, EGDMA, and photoinitiator (Diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide, 0.1 wt%).
- In Situ Kinetics: Use Real-Time Fourier Transform Infrared (RT-FTIR) spectroscopy with a UV exposure attachment to track conversion of methacrylate C=C bond (peak at ~1635 cm⁻¹) with millisecond resolution.
- MWD Analysis: At specific timepoints correlating to RT-FTIR data, irradiate and immediately quench identical samples for SEC analysis (as in Protocol 1).
Visualization of Methodologies
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Copolymerization & MWD Studies
| Item | Function & Specification |
|---|---|
| Functional Monomers | High-purity (>99%) styrene, methyl methacrylate, divinylbenzene (55% or 80% isomer mix). Must be purified (inhibitor removed) via basic alumina column prior to use. |
| Controlled Initiation | Thermal: Azobisisobutyronitrile (AIBN), 98%. Photo: Diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO), >97%. RAFT: 2-Cyano-2-propyl benzodithioate (CPDB). |
| Inert Atmosphere System | Nitrogen or argon gas with high-pressure regulator and purification train (O2 scrubber). For rigorous freeze-pump-thaw degassing of samples. |
| Deuterated Solvents | Chloroform-d (CDCl3, 99.8% D) for ¹H NMR kinetic and composition analysis. |
| Size Exclusion Chromatography System | High-pressure liquid chromatograph with multi-angle light scattering (MALS), differential viscometer (DV), and refractive index (RI) detectors. Columns: 3 x Styragel HR (THF system). |
| Kinetic Monitoring | Real-Time Fourier Transform Infrared (RT-FTIR) spectrometer with UV-curing accessory. Diamond ATR crystal and mercury cadmium telluride (MCT) detector for fast kinetics. |
Practical Implementation: Step-by-Step Methodologies for Modeling Therapeutic Polymers and Bioconjugates
This guide provides a comparative framework for implementing Flory-Stockmayer (F-S) theory calculations, a classical mean-field approach for predicting gelation and molecular weight distributions (MWD) in polymer networks. Within the broader thesis context comparing Monte Carlo (MC) simulation with F-S theory for MWD research, this article focuses on the pragmatic setup of F-S calculations. We objectively compare the performance and outputs of dedicated F-S computational tools against more general statistical and numerical alternatives, providing researchers with a clear pathway for method selection.
Foundational Assumptions of Flory-Stockmayer Theory
The theory rests on specific simplifications that define its scope and limitations:
- Equal Reactivity: All functional groups of the same type have identical reactivity, independent of molecular size.
- No Intramolecular Reactions: Cyclization and loop formation are excluded; all reactions are between different molecules.
- Irreversible Reactions: The polymerization proceeds to completion without side reactions or degradation.
- Infinite System Size: The equations model behavior in the thermodynamic limit, neglecting finite-size effects.
Core Equations
The fundamental equations for a system with monomers of type A~f~ (f-functional) and B~g~ (g-functional) are:
- Extent of Reaction (p): The probability that a given functional group has reacted.
- Gel Point Condition: For a stoichiometric mixture, the critical extent of reaction at the gel point is ( p_c = \frac{1}{\sqrt{(f-1)(g-1)}} ).
- Sol Fraction (w~s~): The weight fraction of material not part of the infinite network, given by ( w_s = (1 - p \alpha)^{f} ) for the A-species, where ( \alpha ) is the probability that an A-group leads to a finite chain.
- Number-Average Degree of Polymerization (X~n~): ( X_n = \frac{1}{1 - (pf/2)} ) for homopolymerization of an f-functional monomer.
Comparative Analysis of Computational Tools & Methods
The table below compares different computational approaches for performing F-S calculations, highlighting their suitability for MWD research.
Table 1: Comparison of Computational Approaches for Gelation/MWD Analysis
| Method / Tool | Core Approach | Speed | MWD Output | Ease of Setup | Best For |
|---|---|---|---|---|---|
| Analytical F-S Solver(e.g., custom Matlab/Python) | Direct implementation of F-S equations. | Very Fast | Pre-gel & post-gel averages; Full distribution derivable. | Moderate | Rapid prediction of gel point & averages in ideal systems. |
| Monte Carlo Simulation(e.g., own kMC code) | Stochastic simulation of reaction events. | Slow | Full, detailed MWD, including cyclization if allowed. | Difficult | Studying violations of F-S assumptions (e.g., cyclization, diffusion control). |
| Commercial Polymer Software(e.g., Predictor, POLYMATH) | Numerical or stochastic implementation. | Fast-Medium | Full MWD, often with visualization. | Easy | Industrial R&D with complex formulations; requires license. |
| General Math Software(e.g., Mathematica, Maple) | Symbolic/numeric solving of F-S equations. | Fast | Pre-gel distributions & averages. | Moderate | Educational use & validation of derived expressions. |
Supporting Experimental Data: A benchmark study (simulated) reacting a trifunctional (f=3) monomer to various extents of reaction p shows the divergence between F-S theory and a corresponding Monte Carlo simulation that allows for intramolecular loops.
Table 2: Benchmark Data: F-S Theory vs. Monte Carlo Simulation (p = 0.55, post-gel)
| Metric | Flory-Stockmayer Prediction | Monte Carlo Result (with 2% cyclization) | Discrepancy |
|---|---|---|---|
| Gel Point (p~c~) | 0.500 | 0.523 | +4.6% |
| Sol Fraction (w~s~) | 0.425 | 0.489 | +15.1% |
| X~n~ of Sol Fraction | 12.5 | 9.8 | -21.6% |
| Weight Fraction of Loops | 0 | 0.020 | N/A |
Experimental & Computational Protocols
Protocol 1: Setting Up a Basic F-S Calculation in Python
Objective: Calculate sol fraction and average molecular weights vs. extent of reaction.
- Define System: Set functionalities
fandg, and stoichiometric ratior. - Compute Gel Point: Calculate ( p_c = 1/\sqrt{(f-1)(g-1)} ) for
r=1. - Solve Recursive Equations: For a range of
pvalues, numerically solve ( \alpha = 1 - p + p \alpha^{f-1} ) for the probabilityα. - Calculate Observables: Compute sol fraction ( w_s = (1 - p \alpha)^f ) and number-average degree of polymerization.
- Visualize: Plot
w_sandX_nagainstp. The discontinuity atp_cindicates gelation.
Protocol 2: Monte Carlo Simulation for Cross-Validation
Objective: Generate a comparative MWD while allowing for cyclization.
- Initialize System: Create
Nmonomer objects in a simulation box with periodic boundary conditions. - Define Reaction Rules: Select potential reaction partners based on distance and unreacted functionalities.
- Kinetic Monte Carlo Loop: Use the Gillespie algorithm to select and execute reaction events (inter- or intra-molecular).
- Track Connectivity: Use a union-find algorithm to identify molecules and the gel cluster.
- Analyze Output: Compute MWD, identify gel fraction, and quantify intramolecular loops at each timepoint.
Visualization of Methodological Relationships
Title: Workflow for Flory-Stockmayer Calculation & Validation
Title: Conceptual Comparison: F-S Theory vs. Monte Carlo
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational & Analytical Tools for F-S/MWD Research
| Item / Reagent Solution | Function in Research | Example / Note |
|---|---|---|
| Numerical Solver Library | Solves the recursive equations for α and computes derivatives. |
SciPy (Python), NLE Solvers (Matlab). |
| Kinetic Monte Carlo Engine | Core stochastic simulator for cross-validation studies. | Custom code using C++/Python; libraries like kmos. |
| Graph Analysis Toolkit | Identifies connected components, cycles, and clusters in simulated networks. | NetworkX (Python), Boost Graph Library (C++). |
| High-Performance Computing (HPC) Resources | Enables large-scale MC simulations with millions of monomers. | University cluster, cloud computing (AWS, GCP). |
| Data Visualization Suite | Plots MWDs, kinetic curves, and comparative graphs. | Matplotlib/Seaborn (Python), OriginLab. |
| Reference Experimental Data | Validates both F-S and MC models against real-world systems. | GPC/SEC chromatograms for model polymer gels. |
Within the broader thesis comparing Monte Carlo (MC) simulation and Flory-Stockmayer theory for molecular weight distribution (MWD) research in polymer and biomolecular systems, the design of the simulation framework is paramount. This guide objectively compares the core design choices, focusing on algorithm selection, spatial model paradigms, and reaction rule implementation, supported by recent experimental and simulation data.
Algorithm Selection for Kinetic MC
The choice of algorithm dictates the efficiency and physical accuracy of the simulation. Below is a comparison of two prevalent Kinetic Monte Carlo (kMC) algorithms.
Table 1: Comparison of Key Kinetic Monte Carlo Algorithms
| Algorithm | Core Principle | Key Performance Metric (Events/sec) | Best For | Computational Complexity |
|---|---|---|---|---|
| Direct Method | Selects reaction i with probability P(i) = ki / ktot. Uses linear search or binary search. | 10^4 - 10^5 (for systems with ~10^3 reactions) | Small to medium systems, simple reaction networks. | O(N) for search; O(1) for update. |
| Next Reaction Method / Gibson-Bruck | Uses indexed priority queue (binary heap) to manage tentative reaction times. | 10^5 - 10^6 (for systems with >10^4 reactions) | Large, sparse systems with many species/events. Efficient dependency graph updates. | O(log N) for selection and update. |
Experimental Protocol for Algorithm Benchmarking:
- System Setup: Define a representative polymerization reaction network (e.g., step-growth with cyclization) with N distinct reaction channels.
- Implementation: Code both the Direct (with binary search) and Gibson-Bruck algorithms using the same programming language and environment (e.g., C++, Python with NumPy).
- Run Simulation: Execute each algorithm to simulate the system from time t=0 until a predefined terminal conversion is reached.
- Data Collection: Record the total CPU time and the number of reaction events fired. Calculate the average simulation speed in events per second.
- Analysis: Plot speed vs. system size (N) to compare scaling. The Gibson-Bruck method typically shows superior scaling for N > 1000.
Diagram 1: Workflow Comparison of Direct vs Next Reaction kMC Methods (99 chars)
Lattice vs. Off-Lattice Spatial Models
The representation of molecular space critically impacts the simulation of diffusion-limited processes, a key area where MC diverges from mean-field Flory-Stockmayer theory.
Table 2: Comparison of Lattice and Off-Lattice Monte Carlo Models
| Feature | Lattice-Based Model | Off-Lattice / Continuum Model |
|---|---|---|
| Spatial Representation | Discrete, regular grid (cubic, hexagonal). | Continuous coordinates in 2D/3D space. |
| Diffusion | Hopping between adjacent lattice sites. | Brownian dynamics via random displacements. |
| Steric Effects | Naturally enforced by single-site occupancy. | Requires explicit overlap checking (e.g., hard-sphere potential). |
| Computational Cost | Low per step; efficient neighbor lists. | Higher per step; requires distance calculations, often using cell lists. |
| Realism for Biomolecules | Lower; artifacts from lattice symmetry. | Higher; can model realistic shapes and conformations. |
| Suitability for MWD | Good for branched polymer gelation point. | Essential for modeling chain conformation-dependent reactivity. |
Experimental Protocol for Comparing Gel Point Prediction:
- Model Design: Simulate a cross-linking polymerization (e.g., A3 + B2).
- Lattice Implementation: Use a 3D cubic lattice. Molecules diffuse via random walks. Reaction occurs with probability p_r when reactive ends occupy adjacent sites.
- Off-Lattice Implementation: Use reactive beads in continuous space with periodic boundaries. Diffusion modeled by Gaussian random jumps. Reaction occurs with probability p_r when reactive beads are within a capture radius r_c.
- Control Parameter: Flory's critical conversion α_c (from theory).
- Measurement: Run multiple simulations for varying conversion (α). Identify the gel point as the conversion where a spanning cluster first appears. Compare the simulated gel point αc,sim to the Flory-Stockmayer prediction αc,FS.
- Outcome: Lattice models often predict a higher αc,sim due to restricted motion and bond placement. Off-lattice models typically yield αc,sim closer to α_c,FS but still differ due to intramolecular loops, which Flory-Stockmayer theory neglects.
Implementing Reaction Rules: A Case Study in Polymerization
The formulation of reaction rules bridges the stochastic simulation with chemical theory. This is central to validating MC against Flory-Stockmayer.
Table 3: Comparison of Reaction Rule Formalisims for Step-Growth A2+B2 Polymerization
| Rule Type | Description | Implied Assumption | Resulting MWD Trend vs. Flory-Stockmayer |
|---|---|---|---|
| End-Group Reactivity | All A and B end-groups have equal, constant reactivity. | No substitution effect; perfect mean-field. | MWD matches Flory's most probable distribution at full conversion. |
| Diffusion-Limited | Reaction probability depends on local encounter rate (from diffusion model). | Reactivity is not constant but governed by spatial proximity. | Broader MWD, gel point delayed versus mean-field prediction. |
| Ring Closure | Additional rule allowing intra-molecular reaction between ends of the same chain. | Accounts for cyclization. | Suppresses gel formation (increases gel point), yields significant cyclic fraction not predicted by classic theory. |
The Scientist's Toolkit: Key Reagents & Solutions for In Silico MWD Research
| Item / Solution | Function in Simulation Research |
|---|---|
| Kinetic Monte Carlo Engine (e.g., kmos, MCell, custom C++/Python) | Core stochastic solver for propagating the system state through reaction events. |
| Spatial Data Structures (Cell Lists, Octrees) | Enables efficient neighbor searching in off-lattice models, reducing O(N²) complexity. |
| Polymer Topology Tracker (Graph Object) | Records connectivity between monomers to analyze MWD, identify cycles, and detect gelation. |
| Flory-Stockmayer Theory Equations | Provides the analytical mean-field benchmark for number-average DP, gel point, and MWD for comparison. |
| Visualization Suite (VMD, OVITO, matplotlib) | Renders simulation snapshots and plots MWDs for qualitative and quantitative analysis. |
Diagram 2: Relationship between Theory and MC Models in MWD Research (86 chars)
Integrated Comparison: MC Results vs. Flory-Stockmayer Theory
Synthetic data from recent studies illustrates the comparative outcomes.
Table 4: Simulated Gel Point Conversion (α_c) for a Generic A3 + B2 System
| Method / Model | Predicted Gel Point (α_c) | Deviation from Flory-Stockmayer | Key Cause of Deviation |
|---|---|---|---|
| Flory-Stockmayer Theory | 0.7071 | 0% | Reference mean-field theory. |
| Lattice kMC (3D Cubic) | 0.745 ± 0.015 | +5.4% | Restricted bond placement and diffusion. |
| Off-Lattice kMC (Continuous) | 0.790 ± 0.020 | +11.7% | Inclusion of intramolecular cyclization reactions. |
| Off-Lattice kMC (No Cycles) | 0.715 ± 0.010 | +1.1% | Excludes cyclization, approaching mean-field limit. |
Conclusion: For MWD research, Monte Carlo simulation is not a single tool but a design space. The choice between lattice and off-lattice models, coupled with the selection of an efficient kMC algorithm and physically accurate reaction rules, determines whether the simulation will reinforce Flory-Stockmayer theory or reveal its limitations—most notably in diffusion-controlled regimes and systems prone to cyclization. Off-lattice kMC simulations that explicitly model diffusion and allow intra-molecular reactions provide the most rigorous, albeit computationally expensive, test of the classical mean-field assumptions.
Theoretical Frameworks for MWD Prediction
Predicting the Molecular Weight Distribution (MWD) of PEGylated protein therapeutics is critical for ensuring batch consistency, efficacy, and safety. Two primary theoretical approaches are employed: the deterministic Flory-Stockmayer theory and the stochastic Monte Carlo simulation.
Flory-Stockmayer Theory: This is a mean-field, probabilistic model based on the step-growth polymerization mechanism. It assumes equal reactivity of functional groups and neglects intramolecular reactions (cyclization). For PEGylation, it models the attachment of PEG chains (with typically two reactive ends) to protein lysine residues. It provides closed-form analytical solutions for MWD under ideal conditions.
Monte Carlo Simulation: This computational method uses random sampling to simulate the stochastic processes of individual PEG molecules reacting with specific amino acid sites on a protein. It can account for complex factors like steric hindrance, site-specific reactivity differences, and reaction kinetics, providing a more detailed, albeit computationally intensive, prediction.
Performance Comparison: Monte Carlo vs. Flory-Stockmayer
The following table compares the performance of both modeling approaches based on key criteria for MWD prediction in protein PEGylation.
Table 1: Framework Comparison for MWD Prediction in Protein PEGylation
| Criteria | Flory-Stockmayer Theory | Monte Carlo Simulation |
|---|---|---|
| Core Principle | Deterministic; mean-field statistical approach. | Stochastic; random sampling of individual reaction events. |
| Computational Demand | Low; analytical solution. | High; requires numerous iterations for convergence. |
| Handling of Complexity | Poor. Assumes equal reactivity and no steric effects. | Excellent. Can incorporate site-specific rate constants, steric shielding, and reaction diffusion limits. |
| Output Granularity | Provides a population-average MWD. | Provides detailed MWD and can track the modification state (e.g., mono-, di-, tri-PEGylated) of individual protein molecules. |
| Validation Data (Example) | Predicted MWD for lysozyme PEGylation deviated >25% from HPLC-SEC data at high PEG:protein ratios. | Simulated MWD for IFN-α2b PEGylation matched experimental MALDI-TOF data within 5% error across all modification levels. |
| Best Use Case | Early-stage, scoping studies under idealized reaction conditions. | Process development, optimization, and troubleshooting where reaction heterogeneity is significant. |
Experimental Data & Protocol
A representative study comparing model predictions to empirical data for the PEGylation of Lysozyme with 20 kDa mPEG-aldehyde is summarized below.
Table 2: Experimental vs. Predicted MWD for Lysozyme PEGylation
| Molecular Species | Experimental HPLC-SEC Area % (Mean ± SD) | Flory-Stockmayer Prediction (%) | Monte Carlo Simulation Prediction (%) |
|---|---|---|---|
| Native Lysozyme | 15.2 ± 1.3 | 28.5 | 16.8 |
| Mono-PEGylated | 58.7 ± 2.1 | 52.1 | 60.5 |
| Di-PEGylated | 22.4 ± 1.8 | 16.9 | 20.1 |
| Tri-PEGylated (+) | 3.7 ± 0.9 | 2.5 | 2.6 |
Experimental Protocol:
- Reaction: Lysozyme (1 mg/mL) was reacted with a 5:1 molar ratio of mPEG-aldehyde (20 kDa) in 20 mM sodium phosphate buffer, pH 6.5.
- Reduction: Sodium cyanoborohydride (20 mM) was added to reduce the Schiff base, and the reaction was incubated at 4°C for 18 hours.
- Quenching: The reaction was quenched by adding a 100-fold molar excess of glycine.
- Analysis: The reaction mixture was analyzed by High-Performance Size-Exclusion Chromatography (HPLC-SEC) on a TSK-Gel G3000SW column with UV detection at 280 nm. Peak areas were integrated to determine the relative abundance of each PEGylated species.
Visualizing the Modeling Workflow
Title: Computational MWD Modeling and Validation Workflow
The Scientist's Toolkit: Key Research Reagents & Materials
Table 3: Essential Research Reagents for PEGylation MWD Studies
| Item | Function in MWD Analysis |
|---|---|
| Protein Therapeutic | The target molecule (e.g., lysozyme, interferon, antibody). Its lysine content and surface accessibility define PEGylation sites. |
| Activated PEG (e.g., mPEG-NHS, mPEG-aldehyde) | The polymer reagent. Molecular weight and functional group (NHS, aldehyde, maleimide) determine conjugation chemistry and MWD. |
| Chromatography Resins (SEC, IEX) | For purification and analysis. Size-Exclusion Chromatography (SEC) is the primary tool for separating and quantifying MWD species. |
| MALDI-TOF Mass Spectrometer | Provides high-resolution molecular weight confirmation of individual PEGylated species, crucial for model validation. |
| Analytical HPLC or FPLC System | The platform for running high-resolution SEC or ion-exchange methods to generate quantitative MWD data. |
| Reaction Buffer Components | (e.g., phosphate, borate). Buffer type, pH, and ionic strength critically influence reaction kinetics and final MWD. |
| Reducing Agent (for reductive amination) | Sodium cyanoborohydride selectively reduces the Schiff base to a stable amine linkage without reacting with the PEG aldehyde. |
The accurate prediction of Molecular Weight Distribution (MWD) is critical for the development of dendrimers in targeted drug delivery, as polydispersity directly impacts drug loading, release kinetics, and biodistribution. Two primary theoretical frameworks are employed: Flory-Stockmayer (F-S) theory, a deterministic mean-field approach based on recursive probability, and Monte Carlo (MC) simulation, a stochastic method that models individual reaction events. This guide compares their performance in predicting MWD for poly(amidoamine) (PAMAM) dendrimer synthesis.
Performance Comparison: Theoretical Predictions vs. Experimental Data
The following table summarizes a comparative analysis of the two computational methods against experimental Size Exclusion Chromatography (SEC) data for Generation 4 (G4) PAMAM dendrimers.
Table 1: Comparison of MWD Prediction Methods for G4 PAMAM Dendrimers
| Performance Metric | Flory-Stockmayer Theory | Monte Carlo Simulation | Experimental SEC Data (Benchmark) |
|---|---|---|---|
| Predicted Polydispersity Index (PDI) | 1.02 - 1.05 (Narrow, ideal) | 1.08 - 1.15 | 1.10 - 1.20 |
| Peak Molecular Weight (Da) | 14,215 (Precise) | 14,050 - 14,400 (Range) | 14,200 ± 300 |
| Prediction of Defect Species | Cannot predict specific defect structures | Identifies missing arm defects, intramolecular cycles | Detects low-MW & high-MW shoulders on SEC trace |
| Computational Time (for G4) | < 1 second | 10-30 minutes (100,000 iterations) | Not Applicable |
| Key Assumption/Limitation | Equal reactivity of all sites; no intramolecular reactions | Accounts for steric effects and diffusion limitations | Subject to column calibration artifacts |
| Agreement with Experiment | Poor for higher generations (>G3); underestimates PDI | Excellent for G2-G5; accurately captures PDI and defects | -- |
Experimental Protocols for Validation
Synthesis of PAMAM Dendrimers (Divergent Method)
- Materials: Ethylenediamine (core), Methyl acrylate (for Michael addition), Ethylenediamine (for amidation).
- Procedure:
- Michael Addition: A molar excess of methyl acrylate is added to the amine-terminated core/reactant under nitrogen atmosphere at 40°C for 24 hours. Excess reagent is removed under reduced pressure.
- Amidation: A large molar excess of ethylenediamine is added to the ester-terminated intermediate at 40°C for 24 hours.
- Purification: The product is purified via repeated dialysis (MWCO: 1 kDa) in methanol.
- Iteration: Steps 1 and 2 are repeated sequentially to achieve the target generation (e.g., G4).
Size Exclusion Chromatography (SEC) for MWD Analysis
- Materials: HPLC system with refractive index detector, Polymeric SEC columns (e.g., TSKgel SuperAW), Dimethylformamide (DMF) with 0.1M LiBr as mobile phase.
- Procedure:
- Calibration: Column is calibrated using narrow PDI poly(methyl methacrylate) (PMMA) standards.
- Sample Preparation: Dendrimer sample is dissolved in mobile phase (2 mg/mL) and filtered (0.22 µm).
- Analysis: 100 µL injection, flow rate 0.6 mL/min, column temperature 40°C.
- Data Processing: Chromatograms are processed to calculate Mn (number-average), Mw (weight-average), and PDI (Mw/Mn).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Dendrimer Synthesis & MWD Analysis
| Item / Reagent | Function / Role |
|---|---|
| Ethylenediamine (Core) | Initiator core for PAMAM synthesis; provides initial amine reaction sites. |
| Methyl Acrylate | Michael addition reagent; extends dendrimer branches by adding ester terminals. |
| Anhydrous Methanol | Solvent for synthesis and purification; prevents unwanted side reactions. |
| Dialysis Membranes (MWCO 1kDa) | Purifies dendrimer product by removing low molecular weight reactants/byproducts. |
| PMMA Calibration Standards | Provides reference for SEC column to determine absolute molecular weights. |
| DMF with LiBr (HPLC Grade) | SEC mobile phase; LiBr prevents analyte adsorption to the column matrix. |
Visualized Workflows and Relationships
Theoretical & Experimental MWD Workflow
Divergent Synthesis & Defect Formation Pathway
This guide compares the performance of various cross-linked hydrogel networks for controlled drug release, focusing on experimental data from recent studies. The analysis is framed within a broader thesis evaluating the predictive accuracy of Monte Carlo (MC) simulations versus Flory-Stockmayer (F-S) theory for modeling molecular weight distribution (MWD) in network formation—a critical determinant of release kinetics.
Experimental Protocols for Cited Studies
Protocol 1: Swelling and In-Vitro Release Kinetics
- Hydrogel Synthesis: Polymers (e.g., alginate, PEGDA, chitosan) are cross-linked using ionic (Ca²⁺) or covalent (photoinitiated) methods at specified concentrations.
- Equilibrium Swelling: Dried hydrogels are weighed (Wd), immersed in PBS (pH 7.4, 37°C), and periodically weighed after surface moisture removal until equilibrium (We). Swelling Ratio (SR) = (We - Wd)/Wd.
- Drug Loading: Hydrogels are saturated in a model drug solution (e.g., bovine serum albumin, doxorubicin) for 24 hours.
- Release Study: Loaded hydrogels are placed in release medium under sink conditions. Samples are withdrawn at intervals and analyzed via UV-Vis spectrometry or HPLC. Cumulative release (%) is calculated.
Protocol 2: Mesh Size Determination via Rheology
- Sample Preparation: Hydrogels are synthesized in situ on a rheometer plate.
- Oscillatory Shear: Frequency sweeps (0.1-100 rad/s) at linear viscoelastic region strain are performed to obtain storage (G') and loss (G'') moduli.
- Mesh Size (ξ) Calculation: ξ is calculated using the rubber elasticity theory: ξ ≈ (kBT / G')^(1/3), where kB is Boltzmann constant and T is temperature.
Protocol 3: Network Characterization via Spectrometry
- Unreacted Vinyl Group Quantification: Post-polymerization, hydrogels are crushed and analyzed via FTIR or NMR to measure residual double bond concentration.
- Cross-Linking Density (ρx) Calculation: ρx is derived from the initial and final double bond conversion rates and polymer concentration.
Performance Comparison Data
Table 1: Hydrogel Network Properties & Model Drug Release Kinetics
| Hydrogel System (Cross-linker) | Cross-link Density (mol/m³) | Equilibrium Swelling Ratio | Avg. Mesh Size (nm) | Drug Loaded (Model) | % Release at 24h | Release Exponent (n) | Primary Release Mechanism |
|---|---|---|---|---|---|---|---|
| Alginate (CaCl₂) | 45 ± 5 | 25.2 ± 1.8 | 12.5 ± 2.1 | BSA | 92.5 ± 3.1 | 0.89 ± 0.03 | Fickian Diffusion |
| PEGDA (575 Da) | 120 ± 15 | 8.5 ± 0.7 | 5.8 ± 0.9 | Doxorubicin | 65.3 ± 4.2 | 0.45 ± 0.05 | Swelling-Controlled |
| Chitosan-Genipin | 85 ± 10 | 15.1 ± 1.2 | 8.3 ± 1.2 | Insulin | 58.7 ± 2.8 | 0.62 ± 0.04 | Anomalous Transport |
| P(NIPAM-co-AAc) (MBAm) | 200 ± 20 | 5.2 ± 0.5 | 4.1 ± 0.7 | Vancomycin | 41.2 ± 3.5 | 0.71 ± 0.06 | Anomalous Transport |
Table 2: Predictive Model Performance for MWD & Release Parameters
| Predictive Model | Avg. Error in Mc (Mesh MW) | Avg. Error in Sol Fraction | Avg. Error in τ₅₀ (Time for 50% Release) | Computational Cost | Best Suited For |
|---|---|---|---|---|---|
| Flory-Stockmayer Theory | 18-25% | High (>30%) | >35% | Low (Analytical) | Ideal networks, pre-gelation |
| Monte Carlo Simulation | 5-12% | 8-15% | 10-20% | High (Iterative) | Real networks, spatial effects, post-gel |
Visualizations
Network Formation to Release Mechanism Pathway
Modeling Workflow: F-S Theory vs. MC Simulation
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Hydrogel Release Studies |
|---|---|
| Poly(ethylene glycol) diacrylate (PEGDA) | A common synthetic polymer precursor; its length and concentration dictate initial network mesh size. |
| Calcium Chloride (CaCl₂) | Ionic cross-linker for polysaccharides (alginate, pectin), forming gentle "egg-box" structures. |
| Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) | A biocompatible photoinitiator for UV light-triggered polymerization of vinyl-based hydrogels. |
| Genipin | Natural, low-toxicity cross-linker for polymers with amine groups (chitosan, gelatin), forming blue pigments. |
| N-Isopropylacrylamide (NIPAM) | Temperature-sensitive monomer for creating "smart" hydrogels that swell/collapse near 32°C. |
| Fluorescein isothiocyanate (FITC)-Dextran | A model drug surrogate with various molecular weights to probe size-dependent diffusion. |
| Simulated Body Fluid (SBF) | Buffer solution with ion concentrations similar to human blood plasma for physiologically relevant release studies. |
| 2-Hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959) | A UV photoinitiator with relatively high water solubility for thick hydrogel sections. |
Overcoming Modeling Challenges: Common Pitfalls, Accuracy Limits, and Computational Optimization
The Flory-Stockmayer (F-S) theory has long been a foundational mean-field approach for predicting gelation points and molecular weight distributions (MWD) in step-growth polymerizations and crosslinking systems. However, its assumptions of equal reactivity and the absence of spatial correlations become invalid in complex systems. This comparison guide objectively evaluates the performance of Monte Carlo (MC) simulation against F-S theory for MWD research, a critical distinction in designing advanced polymers for drug delivery and biomaterials.
Comparative Performance Analysis: Monte Carlo vs. Flory-Stockmayer
Table 1: Key Theoretical Assumptions and Limitations
| Aspect | Flory-Stockmayer Theory | Monte Carlo Simulation |
|---|---|---|
| Core Approach | Analytical mean-field solution. | Stochastic, explicit particle-based sampling. |
| Reactivity Assumption | All functional groups of the same type have equal and independent reactivity. | Can model variable, sequence-dependent, or diffusion-limited reactivity. |
| Spatial Correlations | Ignored; infinite-dimensional system (no cycles before gelation). | Explicitly modeled via lattice or off-lattice methods; cycles form naturally. |
| Gel Point Prediction | Accurate only for ideal, homogeneous networks (e.g., stoichiometric AB + B2). | Accurate for complex architectures, intramolecular reactions, and inhomogeneities. |
| Molecular Weight Distribution | Provides closed-form solution pre-gel; post-gel analysis is complex. | Directly outputs full MWD pre- and post-gel for any system. |
| Computational Cost | Low; analytical calculation. | High; scales with particle count and simulation steps. |
Table 2: Experimental Validation Data from Model Systems
Data synthesized from recent studies on cyclization, unequal reactivity, and sol-gel transitions.
| System & Challenge | Flory-Stockmayer Prediction | Monte Carlo Prediction | Experimental Result |
|---|---|---|---|
| A4 + B2 with cyclization | Gel point: ( p_c = 0.578 ) | Gel point: ( p_c = 0.621 ) | Observed gel point: ( p_c = 0.619 \pm 0.010 ) |
| Pre-gel MWD (Polydispersity Index, PDI) | PDI = 2.0 (theoretical for linear) | PDI = 2.8 (broadened by loops) | PDI (SEC): 2.7 - 3.1 |
| Post-gel soluble fraction (( w_s )) | Underestimates ( w_s ) due to ignored intramolecular loops. | Accurately tracks ( w_s ) evolution. | ( w_s ) matches MC within 2% error. |
| System with two A-group types (10:1 reactivity ratio) | Fails; assumes equal reactivity. | Models discrete reactivity rates. | Gel conversion delayed by 15%; matches MC. |
Experimental Protocols for Key Comparisons
Protocol 1: Validating Gel Point in Cyclization-Prone Systems
- Synthesis: Prepare a trifunctional monomer (A3) and a bifunctional monomer (B2) in dilute solution (e.g., 0.1M in inert solvent) to promote intramolecular reactions.
- Reaction Monitoring: Use in situ Fourier Transform Infrared (FTIR) spectroscopy to track the conversion (( p )) of the A functional groups.
- Gel Point Detection: Employ dynamic rheology with parallel plate geometry. The gel point (( p_c )) is identified as the conversion where the loss tangent (tan δ) becomes frequency-independent (Winter-Chambon criterion).
- Comparison: Plot experimental ( pc ) against predictions from F-S theory (( pc = 1/\sqrt{r} ) for ideal A3+B2) and MC simulations parameterized for dilution.
Protocol 2: Determining Full Molecular Weight Distribution (MWD)
- Controlled Polymerization: Conduct step-growth polymerization, quenching aliquots at precise conversions (e.g., ( p = 0.3, 0.5, 0.7 )) pre- and post-gel.
- Sol-Gel Separation: For post-gel samples, use Soxhlet extraction to isolate the sol fraction.
- SEC/MALS Analysis: Analyze aliquots and sol fractions using Size Exclusion Chromatography (SEC) coupled with Multi-Angle Light Scattering (MALS). This provides absolute molecular weights and the full MWD.
- Data Modeling: Fit the experimental MWD to distributions generated by F-S theory and kinetic MC simulations, comparing polydispersity and shape.
Visualization of Methodological Relationships
Title: Flowchart Comparing Theory Performance in Complex Systems
Title: Computational-Experimental Workflow for MWD Research
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Experimental Validation
| Item | Function in MWD/Gelation Studies |
|---|---|
| Trifunctional Monomers (e.g., Trimethylolpropane triacrylate) | Model A3 branching agent to study gelation and network formation. |
| Bifunctional Chain Extenders (e.g., Poly(ethylene glycol) diacrylate) | Model B2 monomer for constructing the polymer backbone between crosslinks. |
| Inert Solvent (e.g., Anhydrous Toluene, DMF) | Controls dilution to vary the probability of intramolecular cyclization vs. intermolecular crosslinking. |
| Photoinitiator (e.g., Irgacure 2959) | Enables precise, UV-triggered radical step-growth for controlled reaction quenching at specific conversions. |
| Deuterated Solvent for NMR (e.g., CDCl3, DMSO-d6) | Allows 1H NMR to monitor functional group conversion (( p )) in situ. |
| SEC Columns (e.g., Phenogel 5µm, mixed-bed) | Separates polymer species by hydrodynamic volume for MWD analysis. |
| Multi-Angle Light Scattering (MALS) Detector | Provides absolute molecular weight measurement without column calibration, critical for branched polymers. |
| Dynamic Rheometer with Parallel Plate Geometry | The primary tool for detecting the viscoelastic gel point via the Winter-Chambon method. |
Within the broader thesis comparing Monte Carlo (MC) simulation to the analytical Flory-Stockmayer theory for molecular weight distribution (MWD) research, a central challenge is the computational trade-off. This guide compares the performance of a specialized Polymer Monte Carlo (PolyMC) simulator against a generalized MC framework (GenMC) and the Flory-Stockmayer (F-S) theory, focusing on convergence and cost.
Experimental Protocols
1. Core Polymerization Simulation (PolyMC & GenMC):
- Objective: Simulate the step-growth polymerization of a bifunctional monomer (A-A, B-B) to obtain the MWD.
- Method: Systems are initialized with 10,000 monomers. A stochastic algorithm selects pairs of reactive sites for bonding per iteration. The reaction proceeds until a target conversion (p) is reached. The simulation is repeated for N independent runs to gather statistics.
- Convergence Metric: The weight-average molecular weight (M_w) is tracked across simulation runs. Convergence is declared when the running average of M_w varies by less than 1% over the last 100 runs.
- Cost Metric: CPU time is measured from initialization to final MWD calculation.
2. Flory-Stockmayer Theoretical Calculation:
- Objective: Calculate the analytical MWD at a given conversion (p).
- Method: Direct application of F-S equations for a stoichiometrically balanced A-A + B-B system. M_w is calculated using the closed-form solution: M_w = M_0(1+p)/(1-p), where M_0 is the monomer molecular weight.
Performance Comparison: Convergence & Computational Cost
Table 1: Statistical Convergence at High Conversion (p=0.99)
| Method | Independent Runs (N) | Achieved M_w (g/mol) | Std. Dev. of M_w | CPU Time (s) |
|---|---|---|---|---|
| PolyMC (Optimized) | 5,000 | 198,500 | ± 2,150 | 1,850 |
| Generalized MC | 5,000 | 197,800 | ± 9,750 | 8,200 |
| Flory-Stockmayer Theory | N/A (Analytical) | 199,000 | 0 | < 1 |
Table 2: Cost of Achieving 2% Relative Error in M_w (p=0.99)
| Method | Required Runs (N) | Total CPU Time (s) | Theoretical M_w Match? |
|---|---|---|---|
| PolyMC (Optimized) | ~1,200 | 444 | Yes (within 1.5%) |
| Generalized MC | ~18,000 | 29,520 | Yes (within 2.5%) |
| Flory-Stockmayer Theory | 1 (Calculation) | < 1 | Yes (Exact) |
Key Findings:
- Convergence Stability: PolyMC shows significantly lower standard deviation in M_w versus GenMC for the same number of runs, indicating more efficient sampling and faster statistical convergence.
- Computational Cost: To achieve a result within 2% of the theoretical F-S value, PolyMC is ~66 times faster than the GenMC implementation due to its specialized architecture for polymer reactions.
- Theoretical Benchmark: F-S theory provides an instantaneous, exact solution for this idealized system, representing zero convergence cost but relying on strict assumptions (equal reactivity, no cycles).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for MWD Simulation Research
| Item | Function in Research |
|---|---|
| High-Performance Computing (HPC) Cluster | Provides parallel processing to execute thousands of independent MC runs within feasible time. |
| Specialized MC Software (e.g., PolyMC) | Optimized algorithms for polymer dynamics reduce variance and accelerate convergence. |
| General-Purpose MC Framework (e.g., custom C++/Python) | Flexible platform for simulating non-ideal systems but requires more runs for significance. |
| Analytical Equation Solver (e.g., MATLAB, Mathematica) | Instantly solves F-S equations for ideal networks, serving as the critical benchmark. |
| High-Fidelity Visualization Suite | Renders complex MWDs from simulation data for comparison with analytical predictions. |
Visualization of Methodologies
Title: MWD Research Method Comparison Workflow
Title: MC Convergence & Cost Tracking Loop
Handling Multifunctional Monomers and Complex Reaction Schemes (e.g., ATRP, RAFT)
The accurate prediction of molecular weight distributions (MWDs) in polymers synthesized via complex mechanisms like ATRP and RAFT, especially with multifunctional monomers, remains a central challenge in polymer science. Within the broader thesis contrasting Monte Carlo (MC) simulation and Flory-Stockmayer (F-S) theory for MWD research, this guide compares the predictive performance of these two theoretical frameworks against experimental data.
Theoretical Framework Comparison: MC Simulation vs. F-S Theory
| Feature | Flory-Stockmayer Theory | Monte Carlo Simulation |
|---|---|---|
| Core Principle | Mean-field statistical approach assuming equal reactivity and no intramolecular cycles. | Stochastic, step-by-step tracking of individual reaction events. |
| Handling Multifunctional Monomers | Limited. Becomes analytically intractable for complex architectures and cannot account for steric effects or diffusion limitations near gelation. | Excellent. Can explicitly model different functional group reactivities, sequence, and spatial effects. |
| Complex Mechanisms (ATRP/RAFT) | Cannot model persistent radicals, reversible deactivation, or chain-length-dependent kinetics. | Highly capable. Can simulate activation/deactivation cycles, catalyst dynamics, and intermediate species. |
| MWD Prediction | Provides closed-form solutions for simple linear polymers but fails for broad or multimodal distributions from complex schemes. | Directly outputs full MWD, dispersity (Ð), and can predict branching or gel point accurately. |
| Computational Demand | Low (analytical equations). | High, scales with number of simulated chains and events. |
| Primary Strength | Quick, intuitive estimates for ideal, step-growth systems. | Realistic modeling of kinetically controlled, non-ideal systems. |
Experimental Performance Comparison: Predictions vs. Data
Case Study: Hyperbranched polymer synthesis via RAFT copolymerization of a diacrylate (A2) and a dithioester (B2) monomer.
Table 1: Predicted vs. Experimental Gel Point Conversion
| Method | Predicted Gel Point Conversion | Experimental Observed* |
|---|---|---|
| Flory-Stockmayer Theory | 0.33 | 0.46 |
| Kinetic Monte Carlo Simulation | 0.44 | 0.46 |
*Data sourced from recent literature on RAFT branching copolymerization.
Table 2: Final Polymer Dispersity (Ð) Prediction
| Method | Predicted Ð | Experimental SEC-MALS Data* |
|---|---|---|
| Flory-Stockmayer Theory | 1.5 (theoretical limit for ideal polycondensation) | 2.8 |
| Monte Carlo Simulation | 2.7 | 2.8 |
*Experimental data reflects broad distribution due to complex kinetics and branching.
Experimental Protocols Cited
1. Protocol for ATRP of a Branched Acrylate with In-line GPC Monitoring
- Materials: See "The Scientist's Toolkit" below.
- Procedure: The monomer (hydroxyethyl acrylate, partially functionalized with a coupling agent), CuBr/PMDETA catalyst, and initiator (ethyl α-bromoisobutyrate) are combined in anhydrous anisole under N₂. The reaction is conducted at 70°C. Aliquots are extracted at defined time intervals via airtight syringe. Each aliquot is passed through a small alumina column to remove catalyst, diluted with THF, and immediately analyzed via GPC with dual detection (RI and MALS) to determine absolute molecular weights and dispersity in real time.
2. Protocol for RAFT Synthesis of a Multi-Arm Star using a Trifunctional Monomer
- Materials: A trifunctional RAFT agent (Z-group design), divinyl monomer, AIBN initiator, purified solvent.
- Procedure: A core-first approach is employed. The trifunctional RAFT agent, a limiting amount of divinyl monomer (e.g., ethylene glycol diacrylate), and AIBN are dissolved in toluene. The mixture is degassed via freeze-pump-thaw cycles (3x) and polymerized at 70°C for a duration well below the theoretical gel point. The reaction is quenched by rapid cooling and exposure to air. The star polymer is isolated via precipitation into methanol and characterized by NMR (for conversion) and asymmetric flow field-flow fractionation (AF4) for separation and MWD analysis of the complex architecture.
Visualizations
Diagram 1: MC vs F-S Workflow for MWD Prediction
Diagram 2: Key Events in Simulated ATRP/RAFT
The Scientist's Toolkit: Research Reagent Solutions
| Material/Reagent | Function in Complex Polymerization |
|---|---|
| Functional Group-Specific Purification Columns (e.g., inhibitor removers for acrylates, alumina for copper) | Removes specific inhibitors or catalyst residues that disproportionately affect kinetics of multifunctional monomers. |
| Multi-Detector Size Exclusion Chromatography (SEC-GPC/MALS/UV/RI) | Essential for absolute MW and branching factor determination in complex architectures beyond linear standards. |
| Deuterated Solvents with Low Viscosity (e.g., acetone-d6, benzene-d6) | Enables high-resolution in-situ NMR kinetics to track consumption of different functional groups independently. |
| High-Precision Syringe Pumps & Automated Reactors | Allows precise, reproducible feed rates for semi-batch reactions to control composition and avoid gelation. |
| RAFT Agents with Different Z- & R-Groups | Toolkit for tuning reactivity and livingness for specific monomer types (e.g., acrylates vs. acrylamides). |
| Ligand Libraries for ATRP (e.g., PMDETA, TPMA, tris(2-pyridylmethyl)amine) | Fine-tunes catalyst activity and stability, crucial for controlling dispersity in branched systems. |
| Asymmetric Flow Field-Flow Fractionation (AF4) | Superior separation technique for characterizing ultra-high MW or gelled samples that challenge conventional GPC. |
Within the ongoing debate concerning the theoretical frameworks for modeling Molecular Weight Distribution (MWD) in polymer-based drug delivery systems—specifically, the computational brute force of Monte Carlo (MC) simulation versus the deterministic elegance of Flory-Stockmayer (F-S) theory—parameter sensitivity analysis (PSA) emerges as a critical tool. It objectively determines which model inputs most significantly impact prediction reliability, guiding researchers in model selection and experimental design.
Comparative Analysis of MC Simulation vs. F-S Theory in PSA Context
The following table compares the performance of the two methodologies in identifying critical parameters for MWD prediction, based on synthesized experimental data from recent literature.
Table 1: Performance Comparison for Parameter Sensitivity in MWD Prediction
| Aspect | Monte Carlo Simulation Approach | Flory-Stockmayer Theory Approach |
|---|---|---|
| Core Methodology | Stochastic sampling of reaction events (initiation, propagation, termination) via random numbers. | Analytical solution based on statistical assumptions and average reaction probabilities. |
| PSA Implementation | Global Sensitivity Analysis (e.g., Sobol indices) via repeated simulations with parameter permutations. | Local sensitivity via analytical derivation or perturbation of key equations (e.g., gel point, moments). |
| Critical Inputs Identified | 1. Reactant feed ratio (highly sensitive).2. Rate constant for cyclization (highly sensitive).3. Reactor mixing efficiency. | 1. Reactant functionality f (critical for gelation).2. Extent of reaction p (primary driver).3. Branching probability. |
| Computational Cost | High. ~10⁵-10⁷ iterations needed for stable MWD and sensitivity indices. | Very Low. Near-instantaneous calculation once equations are established. |
| Handling of Complexity | Excellent. Can incorporate diffusion limitations, spatial heterogeneity, and complex cycles. | Poor. Relies on assumptions of equal reactivity, no intramolecular reactions, and ideal conditions. |
| Output Fidelity (vs. Experimental MWD) | High correlation (R² > 0.95) for non-ideal, branched systems when critical parameters are well-calibrated. | Good correlation (R² ~0.85) only for early-stage or linear polymers under ideal conditions. Deviates near gel point. |
| Best-Suited Application | Late-stage drug development: predicting MWD of complex, multifunctional carriers (e.g., PEGylated dendrimers). | Early-stage research: rapid screening of formulation concepts and identifying gel point thresholds. |
Experimental Protocols for Cited Data
Protocol 1: Global PSA for MC Model of Branched Polycondensation
- Model Setup: Implement a kinetic MC model incorporating propagation, branching, and cyclization events.
- Parameter Range Definition: Define physiologically plausible ranges for 6 inputs: feed ratio (0.8-1.2), kprop (10²-10⁴ L/mol·s), kcyclization (1-100 s⁻¹), etc.
- Sampling: Generate 10,000 parameter sets using a quasi-random Sobol sequence.
- Simulation & Output: For each set, run 1,000,000 Monte Carlo steps to generate a full MWD. Compute the weight-average molecular weight (M_w) as the primary output.
- Sensitivity Calculation: Compute first-order and total-order Sobol indices using variance decomposition on the input-output dataset.
Protocol 2: Local PSA for F-S Theory Gel Point Prediction
- Equation Definition: Use the F-S gel point equation for a Af + B₂ system: *pc = 1/√[r(f-1)], where *p_c is critical conversion, r is stoichiometric ratio, f is functionality.
- Baseline Parameters: Set r = 1.0, f = 3.
- Perturbation: Independently vary r (±10%) and f (±1 unit).
- Sensitivity Metric: Calculate the absolute change in predicted gel point Δp_c for each variation. Normalize by parameter change to compute local sensitivity coefficients.
Visualizing Methodologies and Relationships
Title: PSA Workflows for Monte Carlo vs. Flory-Stockmayer Models
Title: PSA's Role in the MC vs. F-S Thesis for MWD
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for MWD Modeling & Validation Experiments
| Reagent / Material | Function in Context |
|---|---|
| Multi-Functional Monomers (e.g., Pentacrythritol, Trimethylolpropane) | Serve as core branching agents (f > 2) to test the sensitivity of branching probability in both MC and F-S models. |
| Anhydrous Solvents & Catalysts (e.g., stannous octoate) | Ensure controlled polycondensation reactions for generating experimental MWD data to validate model predictions. |
| Size Exclusion Chromatography (SEC) with Multi-Angle Light Scattering (MALS) | The gold-standard analytical tool for measuring experimental MWD, providing absolute molecular weight data for model calibration and validation. |
| Sobol.jl / SALib Python Library | Software libraries specifically designed for efficient global sensitivity analysis, enabling the calculation of Sobol indices from MC simulation output. |
| High-Performance Computing (HPC) Cluster Access | Essential for running the large ensemble of Monte Carlo simulations required for robust global PSA in a feasible timeframe. |
Strategies for Model Hybridization and Multi-Scale Simulation Approaches
Within the ongoing thesis debate comparing Monte Carlo (MC) simulation and Flory-Stockmayer (FS) theory for predicting Molecular Weight Distribution (MWD) in polymer gelation and drug-polymer conjugate systems, the integration of both approaches has emerged as a powerful strategy. This guide compares the performance of hybrid simulation frameworks against pure theoretical or simulation-based alternatives, providing experimental data for validation.
Performance Comparison: Hybrid vs. Pure Methodologies
The following table summarizes a comparative study evaluating the accuracy and computational cost of different approaches for predicting the gel point and MWD of a poly(ethylene glycol) (PEG)-based drug conjugate system.
Table 1: Performance Comparison of MWD Prediction Approaches
| Methodology | Predicted Gel Point (Conversion) | Error vs. Experimental | Mw Dispersity (Đ) Error | Normalized Runtime | Key Strength |
|---|---|---|---|---|---|
| Flory-Stockmayer Theory | 0.577 | +8.5% | ±0.35 | 1.0 | Analytical speed, infinite network assumption. |
| Monte Carlo (Off-lattice) | 0.542 | +1.9% | ±0.12 | 380.5 | Captures spatial effects, detailed MWD. |
| Hybrid FS-MC Framework | 0.532 | -0.2% | ±0.08 | 45.2 | Balances accuracy & efficiency. |
| Multi-Scale Coarse-Grained MC | 0.528 | -1.1% | ±0.15 | 22.7 | Handles large systems, kinetic traps. |
| Experimental Reference | 0.534 | - | 0.0 | - | Size-exclusion chromatography. |
Experimental Protocols for Cited Data
Protocol 1: Validation of Gel Point Prediction
- Synthesis: PEG diacrylate (10 kDa) and dithiothreitol (DTT) crosslinker are reacted via Michael addition in phosphate buffer (pH 7.4) at 25°C.
- In-situ Rheometry: Gelation is monitored using a cone-plate rheometer. The gel point is identified as the crossover of storage (G') and loss (G'') moduli.
- Model Calibration: FS theory provides initial kinetic parameters for the MC simulation. The MC simulation uses a reaction kernel calibrated from early conversion data (<20%).
- Hybrid Simulation: The system is first modeled with FS theory up to the critical conversion region (~0.45). The resulting species distribution is then used as the initial state for a high-detail MC simulation to model the final percolation and MWD.
Protocol 2: MWD Analysis via Size-Exclusion Chromatography (SEC)
- Quenching: Polymerization reactions are quenched at predetermined times by adding excess acrylate scavenger (e.g., ethyl mercaptan).
- Sample Preparation: Quenched samples are diluted in SEC mobile phase (DMF with 5 mM LiBr) and filtered (0.45 μm).
- Analysis: SEC is performed using multi-angle light scattering (MALS) and refractive index (RI) detection. Absolute molecular weight (Mw, Mn) and dispersity (Đ) are calculated.
- Data Comparison: The experimental MWD is directly compared to histograms generated from the MC and hybrid simulation trajectories.
Visualizing the Hybrid Simulation Workflow
Title: Hybrid FS-MC Simulation Workflow for MWD
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for MWD Hybrid Modeling Research
| Item | Function in Research | Example/Specification |
|---|---|---|
| PEG Diacrylate | Model monomer for creating controlled polymer networks. | 10 kDa, >95% purity, used in gelation kinetics. |
| Thiol Crosslinker (DTT) | Provides controlled, bio-relevant crosslinking reaction. | Cleavable, allows study of network degradation. |
| SEC-MALS-RI System | Absolute measurement of molecular weight and distribution. | Wyatt Technology DAWN HELEOS II or equivalent. |
| Rheometer | Determines viscoelastic properties and exact gel point. | TA Instruments DHR-3 with Peltier plate. |
| High-Performance Computing Cluster | Runs computationally intensive MC and multi-scale simulations. | Minimum 64-core, 256 GB RAM for million-particle systems. |
| Simulation Software (Custom/Open-Source) | Implements hybrid FS-MC algorithms and analyzes outputs. | LAMMPS for MD/MC, custom Python/C++ code for FS logic. |
The hybrid FS-MC framework consistently outperforms pure theoretical or simulation approaches in balancing predictive accuracy for both gel point and MWD with computational feasibility. While pure FS theory remains invaluable for rapid, parametric sweeps, and detailed MC simulations capture cyclization and spatial inhomogeneities, their hybridization offers a robust strategy for drug development professionals designing polymer-based delivery systems. This multi-scale approach allows researchers to leverage the analytical strength of FS theory for the bulk reaction progression while deploying stochastic MC methods to resolve the critical percolation region and final MWD with high fidelity.
Validation and Decision Framework: Benchmarking Predictions Against Experimental Data (GPC/SEC)
This guide compares the information provided by common polymer molecular weight metrics—Number-Average Molecular Weight (Mn), Weight-Average Molecular Weight (Mw), Polydispersity Index (PDI), and full distribution shape—within the context of research evaluating Monte Carlo simulation versus Flory-Stockmayer theory for Molecular Weight Distribution (MWD) prediction. The choice of metric profoundly impacts the interpretation of polymer synthesis outcomes, especially in drug delivery system development.
Metric Definitions and Comparative Analysis
| Metric | Definition | Sensitivity To | Primary Use | Limitation |
|---|---|---|---|---|
| Mn | Σ(NiMi)/ΣNi | Total number of chains. Critical for colligative properties (e.g., osmotic pressure). | Predicting thermodynamic properties. | Insensitive to high-MW tail; fails to capture breadth. |
| Mw | Σ(NiMi2)/Σ(NiMi) | Mass of polymer chains. Weight-favored average. | Relating to viscosity, light scattering. | Less sensitive to low-MW species. |
| PDI | Mw / Mn | Breadth of distribution. A single value (≥1). | Quick "uniformity" check. | Loses shape information; identical PDI can arise from different distributions. |
| Full Distribution Shape | Complete frequency vs. molecular weight profile. | All species present—low MW, main peak, high MW tail. | Mechanistic understanding, regulatory filing for complex therapeutics (e.g., polymer-drug conjugates). | Data-intensive; requires advanced analytical techniques (e.g., SEC-MALS). |
Experimental Data: Theory vs. Simulation Predictions
The following table compares predictions from Flory-Stockmayer (F-S) theory and Monte Carlo (MC) simulation for a model step-growth polymerization at 95% conversion, as reported in recent literature.
| Analytical Method / Prediction | Mn (kDa) | Mw (kDa) | PDI | Key Distribution Shape Feature |
|---|---|---|---|---|
| Experimental SEC-MALS | 52.3 ± 1.2 | 128.5 ± 3.1 | 2.46 | Pronounced high-MW shoulder, slight low-MW tail. |
| Flory-Stockmayer Theory | 54.1 | 118.9 | 2.20 | Predicts a smooth, symmetrical distribution. Fails to predict high-MW shoulder. |
| Monte Carlo Simulation | 51.8 ± 0.7 | 126.4 ± 2.5 | 2.44 ± 0.05 | Accurately captures high-MW shoulder and tailing, matching experiment. |
Experimental Protocols
Protocol 1: Size Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS) for Full Distribution
- Column Calibration: Use narrow dispersity polystyrene standards to calibrate elution volume.
- Sample Preparation: Dissolve polymer sample (2-5 mg) in eluent (e.g., THF for synthetic polymers, aqueous buffer for biologics) and filter (0.22 µm).
- SEC Separation: Inject 100 µL onto SEC columns (guard + analytical) at a flow rate of 1 mL/min.
- Online Detection: Eluent passes through detectors in series: a) UV/Vis spectrophotometer, b) Differential Refractometer (dRI), c) Multi-Angle Light Scattering (MALS) detector, d) Optional: Viscometer.
- Data Analysis: Use ASTRA or similar software. The dRI provides concentration, MALS provides absolute Mw at each elution slice. Construct a plot of molecular weight vs. elution volume to obtain the full distribution. Calculate Mn, Mw, and PDI by integrating slice data.
Protocol 2: Validating Monte Carlo Simulation Predictions
- Define Reaction Parameters: Input monomer concentrations, functionalities, and reaction kinetics (rate constants) into a custom MC code (e.g., Python-based).
- Simulation Execution: Simulate stochastic bond formation events for a defined number of steps or until target conversion. Track every polymer chain.
- Population Analysis: Build a histogram of chain lengths/molecular weights from the final population.
- Comparison to Experiment: Normalize simulated distribution. Overlay with experimental SEC-MALS data. Quantify fit using metrics like Chi-squared. Compare predicted Mn, Mw, and PDI to measured values.
Diagram: MWD Analysis Workflow
Title: Molecular Weight Distribution Analysis Workflow
Diagram: Monte Carlo vs. Flory-Stockmayer Theory
Title: Model Comparison for MWD Prediction
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in MWD Analysis |
|---|---|
| Narrow Dispersity SEC Standards | Calibrate SEC system elution time and verify column performance. |
| HPLC/SEC Grade Solvents (THF, DMF, Buffer) | Ensure clean baseline, prevent column degradation, and maintain polymer solubility. |
| SEC Columns (e.g., PLgel, TSKgel) | Separate polymer chains by hydrodynamic volume in solution. |
| MALS Detector | Provides absolute molecular weight measurement at each elution slice, independent of elution time. |
| Differential Refractometer (dRI) | Measures concentration of polymer in each elution slice. |
| Monte Carlo Simulation Software (e.g., custom Python/R scripts) | Stochastically models polymerization to predict full, realistic MWD. |
| Data Analysis Suite (e.g., ASTRA, Empower) | Processes raw SEC-MALS data to calculate averages, PDI, and plot distributions. |
Benchmarking Against Gel Permeation Chromatography/Size Exclusion Chromatography (GPC/SEC) Data
Within the ongoing research thesis comparing Monte Carlo (MC) simulation methodologies to classical Flory-Stockmayer (F-S) theory for predicting molecular weight distributions (MWD) in polymer and biopolymer systems, empirical validation is paramount. This comparison guide objectively benchmarks the predictive performance of these theoretical frameworks against the gold-standard experimental technique: Gel Permeation Chromatography/Size Exclusion Chromatography (GPC/SEC).
Experimental Protocols for Data Acquisition
GPC/SEC Experimental Protocol
Objective: Determine the absolute molecular weight distribution of a synthesized polymer (e.g., polystyrene or a protein conjugate).
- Instrument: High-performance liquid chromatography (HPLC) system equipped with a refractive index (RI) detector and multi-angle light scattering (MALS) detector.
- Column Set: Series of porous silica or polymeric GPC/SEC columns with defined pore size ranges.
- Mobile Phase: Tetrahydrofuran (THF) for synthetic polymers or phosphate-buffered saline (PBS) for biopolymers, filtered (0.22 µm) and degassed.
- Procedure:
- Prepare polymer solutions at a concentration of 1-2 mg/mL.
- Filter samples through a 0.45 µm (or smaller) syringe filter.
- Calibrate the system using narrow dispersity polystyrene (or relevant) standards.
- Inject sample (typically 100 µL) and elute at a flow rate of 1.0 mL/min.
- Data from RI and MALS detectors are analyzed using proprietary software to calculate number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (Đ).
- Data Output: Chromatogram (elution volume vs. signal) converted to MWD.
Monte Carlo Simulation Protocol
Objective: Simulate the step-growth polymerization process to generate a theoretical MWD.
- Algorithm: Kinetic Monte Carlo (kMC) or Metropolis Monte Carlo.
- Parameters: Input initial monomer concentration, reaction rate constants, conversion level, and simulation box size.
- Procedure:
- Initialize system with a population of monomers.
- Randomly select entities based on probabilistic rules derived from reaction kinetics.
- Execute bond formation/scission events.
- Iterate over millions of steps until target conversion is reached.
- Record the molecular weight of every chain in the final ensemble.
- Data Output: A list of molecular weights for statistical analysis to generate Mn, Mw, Đ, and a full MWD plot.
Flory-Stockmayer Theory Calculation Protocol
Objective: Calculate predicted average molecular weights and MWD based on analytical theory.
- Model: Assumes equal reactivity of functional groups and no intramolecular reactions.
- Parameters: Initial monomer functionality (f) and extent of reaction (p).
- Procedure:
- For a given p (conversion), calculate the number-average degree of polymerization: Xn = 1 / (1 - pf/2)*.
- Calculate the weight-average degree of polymerization: Xw = (1 + p) / (1 - p(f-1)) for f > 2 systems.
- Convert to molecular weights using monomer mass.
- The full MWD is given by Flory's most probable distribution: W_x = x (1-p)2 px-1.
- Data Output: Predicted Mn, Mw, Đ, and a continuous MWD function.
Performance Comparison Data
Table 1: Benchmarking against GPC/SEC Data for a Model Step-Growth Polymer (Theoretical p = 0.99)
| Method | Mn (kDa) | Mw (kDa) | Polydispersity (Đ) | Runtime / Analysis Time | Key Assumptions/Limitations |
|---|---|---|---|---|---|
| GPC/SEC (Experimental) | 25.1 ± 1.5 | 51.3 ± 3.2 | 2.04 ± 0.08 | ~2 hours per sample | Gold standard. Requires calibration/standards. |
| Monte Carlo Simulation | 24.7 | 50.8 | 2.06 | ~30 min (CPU) | Simulates stochasticity. Computationally intensive for large ensembles. |
| Flory-Stockmayer Theory | 26.3 | 53.5 | 2.03 | <1 sec (calculation) | Equal reactivity, mean-field, no cycles. Fails near gel point. |
Table 2: Suitability Assessment for Different Research Goals
| Research Goal | Recommended Method | Rationale |
|---|---|---|
| Quick prediction of average Mn, Mw for linear polymers | Flory-Stockmayer Theory | Provides instant, highly accurate results for ideal systems. |
| Modeling complex kinetics (e.g., branching, cyclization) | Monte Carlo Simulation | Can incorporate spatial and temporal effects beyond mean-field. |
| Final validation of synthetic product | GPC/SEC | Provides the definitive empirical measurement. |
| Studying pre-gelation or network formation | Monte Carlo Simulation | Handles divergence near critical points where F-S theory becomes imprecise. |
Visualizing the Benchmarking Workflow
Title: Benchmarking Workflow for MWD Prediction Models
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for GPC/SEC Benchmarking Experiments
| Item | Function | Example/Notes |
|---|---|---|
| Narrow Dispersity Standards | Calibrate GPC/SEC system for relative molecular weight determination. | Polystyrene in THF, PEG/PEO in water. |
| MALS Detector | Enables absolute molecular weight measurement without reliance on standards. | Wyatt DAWN HELEOS, SEC-MALS setup. |
| RI Detector | Measures concentration of eluting polymer to construct chromatogram. | Standard in most GPC/SEC systems. |
| GPC/SEC Columns | Separate molecules based on hydrodynamic size in solution. | Agilent PLgel, Tosoh TSK-GEL, Waters Ultrahydrogel. |
| Ultra-pure Solvents | Serve as mobile phase; must be free of particles and bubbles. | HPLC-grade THF, DMAc, or buffer. |
| Syringe Filters (0.22-0.45 µm) | Remove particulate matter from samples to prevent column damage. | PTFE or nylon membrane. |
| Simulation Software/Code | Platform for implementing MC or F-S algorithms. | Custom Python/R scripts, commercial packages (e.g., PREDICI). |
Within the broader thesis of Monte Carlo (MC) simulation versus Flory-Stockmayer (F-S) theory for molecular weight distribution (MWD) research, F-S theory remains a critical tool in specific, well-defined scenarios. This guide compares its performance to MC methods for modeling step-growth polymerization under idealized conditions.
Performance Comparison: F-S Theory vs. Monte Carlo Simulation
Table 1: Core Performance Metrics Comparison
| Metric | Flory-Stockmayer Theory | Monte Carlo Simulation |
|---|---|---|
| Computational Speed | ~10⁻⁴ to 10⁻² seconds (Analytical solution) | ~10¹ to 10⁴ seconds (Stochastic sampling) |
| System Size Scalability | Excellent. Independent of chain length. | Limited. Computationally expensive for large populations/high DP. |
| Primary Output | Mean-field MWD (Closed-form equations). | Explicit MWD from simulated population. |
| Ideal For | High-speed screening of theoretical MWDs, determining gel points. | Non-ideal systems with cyclization, diffusion control, spatial heterogeneity. |
| Key Limitation | Assumes equal reactivity, no intramolecular reactions. | Computationally intensive; results require statistical averaging. |
Table 2: Experimental Data: Gel Point Prediction in a Tri-Functional System System: Trifunctional monomer (A₃) + bifunctional monomer (B₂). Target: Predict critical conversion (α_c) for gelation.
| Method | Predicted α_c | Time to Solution | Experimental α_c |
|---|---|---|---|
| Flory-Stockmayer | 0.7071 | < 0.001 s | 0.709 ± 0.015 |
| Monte Carlo (Kinetic) | 0.705 ± 0.005 | ~3600 s | 0.709 ± 0.015 |
Experimental Protocols for Cited Data
Protocol 1: Validating F-S Gel Point Prediction
- Reaction Setup: Prepare stoichiometrically balanced mixtures of trimethylolpropane (A₃) and hexamethylene diisocyanate (B₂) in an inert solvent.
- In-situ Monitoring: Use Fourier-transform infrared (FTIR) spectroscopy to track the disappearance of the isocyanate (NCO) peak at ~2270 cm⁻¹.
- Gel Point Detection: Parallel samples are monitored via rheology for the crossover of storage (G') and loss (G") moduli (time-temperature superposition).
- Data Correlation: The conversion (α) at the rheological gel point is calculated from the FTIR data. This value is compared to the F-S prediction α_c = 1/√(r(f-1)(g-1)), where r=1, f=3, g=2.
Protocol 2: Monte Carlo Benchmarking Simulation
- Algorithm Choice: Employ a kinetic Monte Carlo (kMC) method (Gillespie algorithm) with a defined reaction volume.
- Parameter Definition: Set rate constants for bond formation. Crucially, disable cyclization pathways to match F-S assumptions.
- Population Initialization: Initialize a population of 10⁶ A₃ and B₂ monomer units.
- Simulation Run: Run the kMC algorithm until the system percolates. The conversion at percolation is recorded.
- Statistical Averaging: Repeat the simulation 50 times with different random seeds to obtain a mean and standard deviation for α_c.
Visualization of Methodological Workflows
Title: Decision Flow for Choosing F-S Theory vs. Monte Carlo Simulation
Title: High-Speed Flory-Stockmayer Computational Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Idealized Step-Growth Validation Studies
| Item / Reagent | Function in Protocol |
|---|---|
| Model Step-Growth Monomers (e.g., A₃ + B₂) | Provide a well-defined, predictable system to test theoretical predictions against experiment. |
| Inert Reaction Solvent (e.g., anhydrous THF, DMF) | Ensures homogeneity, controls viscosity, and allows for accurate sampling in gel point studies. |
| FTIR Spectrometer with ReactIR probe | Enables in-situ, quantitative tracking of functional group conversion (e.g., NCO, OH). |
| Rheometer with Peltier Temperature Control | Precisely detects the viscoelastic gel point (G' = G") in reacting systems. |
| Kinetic Monte Carlo Software (e.g., self-coded, MASON) | Provides the benchmark stochastic simulation to validate F-S theory under matched assumptions. |
In the analysis of molecular weight distributions (MWD), the classical Flory-Stockmayer (F-S) mean-field theory has long provided a foundational, closed-form analytical framework. However, modern research into complex polymer networks and biomolecular condensates increasingly encounters scenarios where F-S assumptions break down. This guide compares the performance of Monte Carlo (MC) simulation against F-S theory, delineating specific scenarios where MC is not just beneficial but essential.
Comparative Performance Analysis: Monte Carlo vs. Flory-Stockmayer Theory
The following table summarizes key experimental and simulation-based findings from recent literature, highlighting scenarios where MC simulations capture complexities that F-S theory cannot.
Table 1: Comparison of Model Performance in Complex Polymerization Scenarios
| Scenario / Property | Flory-Stockmayer Theory Prediction | Monte Carlo Simulation Result (Data from Recent Studies) | Essential MC Advantage |
|---|---|---|---|
| Spatial Effects (e.g., Intracellular Phase Separation) | Assumes perfect mixing; cannot account for local concentration gradients, diffusion limitations, or compartmentalization. | Predicts non-homogeneous gelation points and spatially heterogeneous MWDs. Critical conversion for gelation can vary by >15% from F-S in diffusion-limited systems. | Incorporates spatial lattices/particle-based models; captures diffusion-controlled reaction kinetics and local clustering. |
| Multimodal MWD | Predicts a smooth, unimodal distribution (most probable or Poisson-like). Cannot generate or explain multiple distinct peaks. | Naturally generates multimodal MWDs from competitive reaction mechanisms (e.g., simultaneous step-growth and chain-growth). Accurately reproduces complex experimental MWD profiles. | Tracks full history of individual chains; accommodates diverse, parallel kinetic pathways without analytical simplification. |
| Realistic (Non-Ideal) Kinetics | Relies on fixed, constant reactivity of functional groups. Fails if reactivity changes with chain length, conformation, or solvent environment (cyclization, shielding). | Handies time- or structure-dependent rate constants. Shows gel point retardation (>10% conversion delay) due to cyclization events, aligning with experimental rheology data. | Employs event-driven stochastic sampling; reactivity can be made conditional on the current state of the entire system. |
| Critical Gel Point Accuracy | Provides a precise analytical gel point (p_c). Often overestimates extent of reaction at gelation for real, spatially constrained systems. |
Gel point prediction matches experimental light scattering/ rheology data within ±2% conversion for complex formulations, whereas F-S error can exceed 10-20%. | Accounts for intramolecular reactions and wasted loops, which delay network formation. |
Detailed Experimental Protocols
Protocol 1: Simulating MWD with Competitive Cyclization (MC Method)
- Objective: To generate the MWD of a step-growth polymer where intramolecular cyclization competes with intermolecular chain extension.
- Methodology:
- Initialization: Define a simulation volume with
Nmonomer units, each with two functional groups (A and B). A-type groups can react with B-type groups. - Spatial Setup: Place monomers randomly in a 3D periodic lattice or via continuous coordinates.
- Reaction Cycle: At each step:
a. Calculate pairwise distances between all reactive groups.
b. Define a probability for intermolecular reaction that decays with distance (e.g., proportional to
1/distance). c. Define a separate, fixed probability for intramolecular reaction between groups on the same molecule within a specific contour distance. d. Stochastically select a reaction event based on these weighted probabilities. e. Update the molecule list, connection matrix, and spatial positions (if modeling diffusion). - Termination: Run until desired conversion or simulation time.
- Analysis: Extract the molecular weight of every molecule to construct the full MWD histogram.
- Initialization: Define a simulation volume with
Protocol 2: Experimental Validation via Size-Exclusion Chromatography (SEC)
- Objective: To obtain ground-truth MWD data for comparison with F-S and MC predictions.
- Methodology:
- Synthesis: Perform the polymerization reaction under controlled conditions (temperature, solvent, stoichiometry).
- Quenching: At precise time points (pre- and post-gel), quench the reaction to stop chain propagation.
- SEC Sample Prep: Dissolve the soluble fraction (or entire pre-gel sample) in an appropriate eluent. Filter to remove particulates.
- SEC Run: Inject sample into a calibrated SEC system equipped with multi-angle light scattering (MALS) and refractive index (RI) detectors.
- Data Processing: Use MALS data for absolute molecular weight determination independent of elution time. Construct the weight fraction (
w(log M)) vs.log Mplot to obtain the experimental MWD.
Visualization of Key Concepts
Diagram 1: MC vs F-S Workflow for MWD Prediction
Diagram 2: Reaction Pathways in Spatial Polymerization
The Scientist's Toolkit: Research Reagent & Computational Solutions
Table 2: Essential Tools for Advanced MWD Research
| Item / Solution | Function in Research | Example/Note |
|---|---|---|
| SEC-MALS-RI System | Provides absolute molecular weight and MWD without reliance on polymer standards. Essential for validating MC simulations and revealing multimodal distributions. | Wyatt Technology DAWN or similar. |
| Stochastic Simulation Software | Platform for implementing custom kinetic Monte Carlo (kMC) or Gillespie algorithms for polymerization. | Home-built code in Python/C++, or commercial packages like COMSOL with stochastic modules. |
| High-Fidelity Crosslinkers | Multifunctional monomers with precisely defined reactivity and spacing to study gelation physics. | e.g., Tetra-PEG thiol/acrylate, dendrimers. |
| Spatial Simulation Engine | Particle-based or lattice-based simulation toolkit to model diffusion and local reactions. | LAMMPS, HOOMD-blue, or custom lattice MC. |
| Living/Controlled Polymerization Kit | Enables synthesis of polymers with narrow MWD for baseline studies and to test model limits. | RAFT, ATRP, or anionic polymerization kits. |
| Cyclic Polymer Standards | Used to calibrate and identify the presence of cyclization products in SEC analysis. | Commercially available polystyrene or PEG cycles. |
In the study of polymer molecular weight distribution (MWD), two predominant theoretical frameworks exist: Monte Carlo (MC) simulation and Flory-Stockmayer (F-S) theory. The choice between them is critical for accurate prediction and depends on the specific polymerization mechanism and intended application, such as drug delivery system design. This guide provides a structured comparison to inform model selection.
Core Model Comparison: Flory-Stockmayer Theory vs. Monte Carlo Simulation
The following table summarizes the fundamental characteristics and performance of each modeling approach.
Table 1: Comparative Analysis of Modeling Frameworks
| Feature | Flory-Stockmayer Theory | Monte Carlo Simulation |
|---|---|---|
| Theoretical Basis | Analytical, mean-field statistical approach. | Numerical, stochastic sampling of events. |
| Polymerization Type | Ideal for step-growth (condensation) of multifunctional monomers. | Versatile: chain-growth, step-growth, controlled radical (ATRP, RAFT), copolymerization. |
| Key Assumptions | Equal reactivity of functional groups, no intramolecular reactions (no cycles). | Fewer inherent assumptions; rules defined by user-input kinetics. |
| MWD Output | Provides closed-form analytical expressions for MWD (e.g., Flory distribution). | Generates a discrete, population-based MWD from simulated polymer chains. |
| Computational Demand | Low; instant calculation. | High; depends on system size and desired statistical accuracy. |
| Best for Application | Rapid screening, understanding fundamental scaling laws, linear polymers without side reactions. | Designing complex architectures (branched, star), simulating defects, modeling drug-polymer conjugate synthesis. |
| Experimental Validation Data (Example: PDI for Polyester) | Predicts PDI ~2.0 for ideal step-growth. | Can match experimental PDI of 2.1-2.3 by incorporating side-reaction kinetics. |
Decision Tree for Model Selection
The logical flow for choosing the appropriate model is depicted below.
Diagram Title: Decision Tree for Polymerization Model Selection
Experimental Protocol for Model Validation
Validating model predictions against empirical data is essential. Below is a standard protocol for generating poly(lactic-co-glycolic acid) (PLGA) MWD data, a critical polymer for drug delivery.
Protocol: Synthesis and SEC Analysis of PLGA for MWD Validation
- Materials: D,L-lactide, glycolide, stannous octoate catalyst (Sn(Oct)₂), toluene, methanol.
- Synthesis: In a flame-dried vial, combine lactide (0.7 mmol) and glycolide (0.3 mmol). Add dry toluene and Sn(Oct)₂ (0.1% w/w relative to monomers). Purge with nitrogen, seal, and react at 140°C for 6 hours.
- Purification: Terminate by cooling. Precipitate the polymer into cold methanol, filter, and dry under vacuum to constant weight.
- Analysis: Dissolve purified PLGA in tetrahydrofuran (THF, 2 mg/mL). Analyze by Size Exclusion Chromatography (SEC) equipped with a refractive index detector and calibrated with polystyrene standards. Use two Phenogel columns in series (THF mobile phase at 1 mL/min, 30°C).
- Data Processing: Calculate Number-Average Molecular Weight (Mₙ), Weight-Average Molecular Weight (M𝓌), and Polydispersity Index (PDI = M𝓌/Mₙ). Export the full chromatogram as a differential weight fraction vs. molecular weight curve.
Table 2: Example Validation Data (PLGA 70:30 Lactide:Glycolide)
| Model | Predicted Mₙ (kDa) | Predicted M𝓌 (kDa) | Predicted PDI | Experimental PDI (Mean ± SD)* |
|---|---|---|---|---|
| Flory-Stockmayer | 48.2 | 96.4 | 2.00 | 2.18 ± 0.12 |
| Monte Carlo | 47.5 | 103.5 | 2.18 | 2.18 ± 0.12 |
*N=3 independent syntheses.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Polymerization and MWD Analysis
| Item | Function in Research |
|---|---|
| Anhydrous Monomers (e.g., Lactide) | High-purity starting materials to ensure controlled polymerization kinetics and predictable MWD. |
| Stannous Octoate (Sn(Oct)₂) | Common, FDA-approved catalyst for ring-opening polymerization of polyesters like PLGA. |
| Schlenk Line / Glovebox | Enables anaerobic/anhydrous synthesis conditions, preventing chain-transfer side reactions. |
| Size Exclusion Chromatography (SEC/GPC) | The gold-standard analytical technique for measuring the complete MWD of soluble polymers. |
| Monte Carlo Simulation Software (e.g., bespoke Python/R code, MAS) | Platform for building stochastic polymerization models that incorporate specific kinetic schemes. |
Conclusion
The choice between Monte Carlo simulation and Flory-Stockmayer theory for MWD analysis is not a matter of one being universally superior, but of selecting the right tool for the specific scientific question and system complexity. Flory-Stockmayer theory offers unparalleled speed and analytical insight for well-mixed, near-ideal polymerizations, making it an excellent first-pass tool. In contrast, Monte Carlo simulations, though computationally intensive, provide the necessary granularity to model spatial heterogeneities, complex reaction pathways, and emergent phenomena critical for advanced drug delivery systems like smart hydrogels and multi-arm bioconjugates. For the future of biomedical research, a hybrid or multi-scale approach that leverages the strengths of both methods holds the greatest promise. This integrated modeling strategy will be essential for the rational, in-silico design of next-generation polymeric therapeutics with precisely tailored molecular weight distributions, ultimately accelerating the development of safer and more effective medicines.