Harnessing the EPR Effect: A Comprehensive Guide to Polymeric Nanoparticles for Targeted Drug Delivery
This article provides a detailed exploration of the Enhanced Permeability and Retention (EPR) effect as the cornerstone of passive tumor targeting for polymeric nanoparticles.
Harnessing the EPR Effect: A Comprehensive Guide to Polymeric Nanoparticles for Targeted Drug Delivery
Abstract
This article provides a detailed exploration of the Enhanced Permeability and Retention (EPR) effect as the cornerstone of passive tumor targeting for polymeric nanoparticles. Tailored for researchers and drug development professionals, it covers fundamental principles, advanced synthesis and functionalization methodologies, key challenges in clinical translation with optimization strategies, and critical evaluation through pre-clinical models and comparative analysis with other nanocarriers. The content synthesizes current research to offer a practical roadmap from nanoparticle design to potential clinical application, addressing both the promise and the pitfalls of EPR-based nanomedicines.
The EPR Effect Decoded: The Foundational Science Behind Passive Tumor Targeting
The Enhanced Permeability and Retention (EPR) effect is a foundational concept in oncology drug delivery, describing the phenomenon by which macromolecular agents and nanoparticles selectively accumulate in tumor tissues. This whitepaper, framed within broader thesis research on EPR and polymeric nanoparticles, details the core principles, historical evolution, and critical methodologies for its study, aimed at researchers and drug development professionals.
Historical Context and Evolution
The EPR effect was first described by Matsumura and Maeda in 1986. Their seminal work observed that macromolecules (e.g., the polymer-drug conjugate SMANCS) leaked from the aberrant tumor vasculature and were retained due to poor lymphatic drainage. This was a paradigm shift from the belief that tumors had impermeable vessels. Subsequent research has revealed significant heterogeneity in the EPR effect across tumor types, locations, and individual patients, prompting strategies for its augmentation.
Core Pathophysiological Principles
The EPR effect arises from distinct anatomical and pathophysiological features of solid tumors.
2.1 Tumor Vasculature Abnormalities Rapid angiogenesis in tumors leads to vessels that are disorganized, dilated, and leaky. Key mediators include Vascular Endothelial Growth Factor (VEGF), bradykinin, and prostaglandins, which enhance vascular permeability. Endothelial gaps (fenestrations) ranging from 100 nm to 2 µm facilitate extravasation.
2.2 Deficient Lymphatic Drainage Tumors often lack a functional lymphatic system, which, combined with the dense extracellular matrix, impedes the clearance of extravasated macromolecules, leading to their prolonged retention.
Quantitative Characterization of the EPR Effect
The efficiency of the EPR effect is quantified using key pharmacokinetic and biodistribution parameters. The following table summarizes critical metrics from recent studies (2020-2024) utilizing polymeric nanoparticles.
Table 1: Quantitative Metrics for EPR Effect Evaluation in Preclinical Models
| Parameter | Typical Range/Value | Measurement Technique | Significance |
|---|---|---|---|
| Tumor Accumulation (%ID/g)* | 3-10% ID/g (Passive) | Radioisotope tracing, NIRF imaging | Measures total dose delivered to tumor. |
| Tumor-to-Normal Tissue Ratio | 3:1 to 10:1 (Varies widely) | Ex vivo biodistribution, PET/CT | Indicator of targeting selectivity. |
| Plasma Half-life (t₁/₂) | 10-100 hours (Polymer-coated NPs) | Blood sampling, pharmacokinetic analysis | Longer circulation enhances EPR. |
| Average Pore Size (Cut-off) | 200-1200 nm | Perfusion with dextrans of varying sizes | Defines size limit for extravasation. |
| Interstitial Fluid Pressure (IFP) | 20-100 mmHg (vs. ~0 in normal) | Micropressure transducer | High IFP can hinder uniform penetration. |
*%ID/g = Percentage of Injected Dose per gram of tissue.
Key Experimental Protocols for EPR Evaluation
4.1 Protocol: Assessing Macromolecular Accumulation via Fluorescence Imaging Objective: To visualize and quantify the extravasation and retention of nanocarriers in a subcutaneous tumor model. Materials: Murine tumor model (e.g., CT26, 4T1), fluorescently-labeled polymeric nanoparticles (e.g., DiR-labeled PLGA-PEG NPs, 80-150 nm), in vivo imaging system (IVIS). Procedure:
- Nanoparticle Administration: Inject NPs intravenously via the tail vein (dose: 5 mg/kg nanoparticle content).
- In Vivo Imaging: Anesthetize mice at predetermined time points (1, 4, 24, 48 h). Acquire fluorescence images (excitation/emission appropriate for dye).
- Ex Vivo Biodistribution: Euthanize mice at terminal time point (e.g., 48 h). Excise tumors and major organs. Image organs ex vivo to quantify fluorescence intensity.
- Data Analysis: Convert fluorescence signals to %ID/g using a standard curve from spiked control tissue. Calculate tumor-to-muscle and tumor-to-liver ratios.
4.2 Protocol: Measuring Tumor Vascular Permeability Objective: To quantify the permeability of tumor vasculature using the Evans Blue (EB) dye method. Materials: Evans Blue dye (0.5% w/v in saline), formamide, spectrophotometer. Procedure:
- Dye Administration: Inject EB (4 mL/kg) intravenously and allow circulation for 30 minutes.
- Perfusion: Sacrifice mouse and perfuse systemically with saline via the left ventricle to remove intravascular dye.
- Tumor Harvest & Extraction: Weigh the excised tumor, homogenize in formamide (1 mL/100 mg tissue). Incubate at 60°C for 24h to extract EB.
- Quantification: Centrifuge homogenate. Measure absorbance of supernatant at 620 nm. Calculate µg EB/mg tissue from a standard curve.
Diagram: VEGF-Mediated Pathway in EPR Effect
Title: VEGF Signaling Cascade Leading to Vascular Permeability
Diagram: Experimental Workflow for EPR Quantification
Title: Preclinical Workflow for NP Accumulation and EPR Study
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagent Solutions for EPR and Polymeric Nanoparticle Research
| Item | Function & Application |
|---|---|
| PLGA-PEG Copolymer | Forms the core-shell matrix of "stealth" nanoparticles, providing biodegradability (PLGA) and prolonged circulation (PEG). |
| Cy5.5, DiR, ICG Dyes | Near-infrared fluorophores for labeling nanoparticles to enable deep-tissue in vivo and ex vivo fluorescence imaging. |
| Recombinant VEGF | Used in in vitro assays (e.g., endothelial monolayer permeability) to simulate tumor angiogenesis signaling. |
| Evans Blue Dye | A classic albumin-binding dye used to visually and spectrophotometrically quantify vascular leakage. |
| Matrigel | Basement membrane extract used for establishing orthotopic or cell-derived subcutaneous tumor models. |
| D-Luciferin | Substrate for bioluminescence imaging (BLI); used in tumor cells expressing luciferase for growth monitoring. |
| Dextrans (FITC-labeled, various MW) | Polysaccharide probes of defined molecular weight/size used to characterize vascular pore cut-off size. |
| Protease Inhibitor Cocktail | Essential for homogenizing tumor tissue for protein or drug analysis without degrading the target analyte. |
This technical whitpaper examines the dual pathophysiological pillars enabling the Enhanced Permeability and Retention (EPR) effect: aberrant tumor vasculature and dysfunctional lymphatic drainage. Framed within contemporary research on polymeric nanoparticle drug delivery, this guide details the molecular mechanisms, quantitative benchmarks, experimental methodologies, and research tools essential for exploiting this phenomenon in oncology therapeutics.
The Enhanced Permeability and Retention (EPR) effect, a cornerstone of modern tumor-targeted drug delivery, is predicated on two distinct yet concurrent vascular abnormalities specific to solid tumors: 1) hyperpermeable blood vasculature and 2) deficient lymphatic drainage. This creates a "leaky and retain" environment, facilitating the selective extravasation and accumulation of macromolecules and nanoparticles. Understanding the pathophysiological basis is critical for designing effective polymeric nanocarriers.
Pathophysiology of Tumor Vasculature Leakiness
Molecular Drivers of Angiogenesis and Hyperpermeability
Tumor vasculature is characterized by immature, chaotic angiogenesis driven by a hypoxic microenvironment. Key signaling pathways involve:
- VEGF/VEGFR2 Axis: Vascular Endothelial Growth Factor (VEGF-A) is the primary mediator, binding to VEGFR2 on endothelial cells. This activates downstream pathways (e.g., Src, FAK, eNOS) leading to altered cytoskeletal organization, disrupted adherens junctions (VE-cadherin), and increased vesiculo-vacuolar organelle (VVO) formation.
- Angiopoietin-Tie2 System: Ang-2, expressed in tumor endothelium, antagonizes the stabilizing Ang-1/Tie2 interaction, promoting vessel destabilization and enhancing VEGF sensitivity.
- Inflammatory Mediators: Tumor-secreted bradykinin, prostaglandins, and matrix metalloproteinases (MMPs) further degrade the basement membrane and increase permeability.
Diagram 1: VEGF-driven signaling pathways in tumor vascular leakiness.
Quantitative Characterization of Leakiness
Vessel leakiness is heterogeneous, both between tumor types and within a single tumor. Key measurable parameters include:
Table 1: Quantitative Parameters of Tumor Vasculature Leakiness
| Parameter | Normal Vasculature | Tumor Vasculature | Measurement Technique |
|---|---|---|---|
| Pore Cut-off Size | 5-7 nm | 200 nm - 1.2 μm | Fluorescent dextran/bead accumulation |
| Permeability Coefficient (P) | ~0.5-2 x 10⁻⁷ cm/s | 10-50 x 10⁻⁷ cm/s | Evans Blue Dye, Radiolabeled Albumin |
| Vessel Diameter | Consistent, hierarchical | Irregular, dilated | Immunohistochemistry (CD31) |
| Pericyte Coverage | High (>70%) | Low, loose attachment (<30%) | IHC (α-SMA, NG2, CD31 co-staining) |
Pathophysiology of Impaired Lymphatic Drainage
Mechanisms of Lymphatic Dysfunction
While angiogenesis is rampant, lymphangiogenesis in tumors is often dysfunctional or absent, leading to poor clearance of interstitial fluid and macromolecules.
- Mechanical Compression: Rapid tumor cell proliferation physically compresses and collapses initial lymphatic vessels.
- Lymphatic Maturation Defects: Newly formed lymphatic vessels (driven by VEGF-C/VEGFR3) are often non-functional, lacking proper connections and valve structures.
- Increased Interstitial Pressure: The combination of leaky inflow and blocked outflow leads to elevated interstitial fluid pressure (IFP), which equilibrates in the tumor core, paradoxically hindering convection.
Quantitative Characterization of Drainage Impairment
Table 2: Parameters of Tumor Lymphatic Function and Interstitial Environment
| Parameter | Normal Tissue | Tumor Tissue | Measurement Technique |
|---|---|---|---|
| Interstitial Fluid Pressure (IFP) | 0 to -3 mmHg | 5-40 mmHg (high in core) | Wick-in-needle, Micropuncture |
| Lymphatic Density | Organized network | Low, peripheral only | IHC (LYVE-1, Podoplanin) |
| Collagen Content & Structure | Organized fibrils | Dense, cross-linked, disordered | Masson's Trichrome, SHG imaging |
| Hyaluronan Content | Moderate | Often highly elevated | Histochemistry, ELISA |
Diagram 2: Relationship between leakiness, poor drainage, and the EPR effect.
Experimental Protocols for EPR Characterization
Protocol: Quantifying Vascular Permeability via Evans Blue Dye
Objective: To measure the extravasation of albumin-bound dye as an index of vascular permeability. Materials: Evans Blue dye, phosphate-buffered saline (PBS), formamide, saline, spectrophotometer. Procedure:
- Inject Evans Blue dye (30 mg/kg, i.v.) into tumor-bearing mouse model.
- After 30 minutes, perfuse the mouse extensively with saline via the left ventricle to clear intravascular dye.
- Excise tumor and homogenize in formamide (1 mL/100 mg tissue).
- Incubate at 60°C for 24 hours to extract dye.
- Centrifuge homogenate and measure supernatant absorbance at 620 nm.
- Calculate dye content from a standard curve. Express as µg dye per g tumor tissue.
Protocol: Assessing Nanoparticle Accumulation (EPR Effect)
Objective: To visualize and quantify the tumor accumulation of fluorescent polymeric nanoparticles. Materials: Fluorescently labeled (e.g., Cy5.5, DiR) polymeric nanoparticles, in vivo imaging system (IVIS), tissue homogenizer, fluorescence spectrometer. Procedure:
- Administer fluorescent nanoparticles (5-10 mg/kg, i.v.) to tumor-bearing mice.
- At predetermined time points (e.g., 1, 4, 24, 48 h), image mice using IVIS to assess whole-body biodistribution.
- Euthanize mice, collect tumors and major organs (liver, spleen, kidney, heart, lung).
- Weigh tissues and either (a) image ex vivo using IVIS, or (b) homogenize in PBS and measure fluorescence intensity.
- Calculate % injected dose per gram of tissue (%ID/g) using a standard curve of known nanoparticle concentrations.
Protocol: Measuring Interstitial Fluid Pressure (IFP)
Objective: To determine IFP using the wick-in-needle technique. Materials: Wick-in-needle apparatus (hypodermic needle with nylon fiber), pressure transducer, saline-filled tubing, data acquisition system. Procedure:
- Anesthetize the animal and place the tumor side up.
- Insert the wick-in-needle connected to the pressure transducer into the tumor core.
- Allow pressure to stabilize (2-3 minutes).
- Record the mean stable pressure reading. Compare to a contralateral normal tissue site.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for Studying Tumor Vascular Pathology
| Reagent/Material | Supplier Examples | Primary Function in Research |
|---|---|---|
| Recombinant VEGF-A | PeproTech, R&D Systems | Positive control for inducing endothelial permeability in vitro. |
| VEGFR2 (Flk-1) Inhibitor (SU5416) | Selleckchem, Tocris | To pharmacologically block VEGF signaling and validate pathway specificity. |
| Fluorescent Dextrans (various MW) | Thermo Fisher, Sigma-Aldrich | Tracer molecules of defined size to quantify vascular pore cut-off size in vivo. |
| Anti-CD31 Antibody | BioLegend, Abcam | Endothelial cell marker for immunohistochemistry to visualize vessel density and morphology. |
| Anti-α-SMA Antibody | Sigma-Aldrich, Abcam | Marker for pericytes; used with CD31 to assess pericyte coverage. |
| Anti-LYVE-1 Antibody | R&D Systems, Abcam | Lymphatic endothelial cell marker to assess lymphatic vessel density and location. |
| Matrigel | Corning | Basement membrane extract for in vitro tube formation assays and in vivo plug assays for angiogenesis. |
| Near-Infrared (NIR) Fluorescent Dyes (Cy5.5, DiR) | Lumiprobe, BioLegend | For labeling polymeric nanoparticles for in vivo and ex vivo tracking studies. |
| Evans Blue Dye | Sigma-Aldrich | Classic dye that binds serum albumin for quantifying macromolecular permeability. |
Implications for Polymeric Nanoparticle Design
The quantitative understanding of pore size (Table 1) directly informs nanoparticle design. Optimal EPR-mediated accumulation is achieved with particles sized between 50-200 nm, small enough to extravasate but large enough to avoid rapid renal clearance. Surface charge must be near-neutral to slightly negative to minimize non-specific interactions. Furthermore, strategies to modulate the tumor microenvironment (e.g., reducing IFP with angiotensin blockers, normalizing vasculature with anti-angiogenics) are being explored to potentiate the EPR effect. This pathophysiological basis remains fundamental to advancing the next generation of tumor-targeted polymeric nanotherapeutics.
The Enhanced Permeability and Retention (EPR) effect remains a cornerstone principle in the targeted delivery of nanomedicines to solid tumors. It exploits the pathological anatomy of tumor vasculature—characterized by leaky endothelial gaps—and impaired lymphatic drainage. While the EPR effect provides a foundational rationale, its effective harnessing for polymeric nanoparticle (PNP) drug delivery is not passive. This whitepaper, framed within ongoing research into optimizing PNPs for oncology, details the three critical physicochemical determinants that dictate a nanoparticle's journey from injection to tumor deposition: particle size, surface charge (zeta potential), and hydrophobicity. Mastery of these parameters is essential for translating the theoretical promise of the EPR effect into clinically efficacious therapies.
Quantitative Analysis of Key Determinants
The following tables synthesize current experimental data on the optimal ranges and functional impacts of each parameter.
Table 1: Impact of Particle Size on EPR Efficacy and Biodistribution
| Size Range (nm) | Primary Effect on Vasculature | Tumor Penetration Depth | Primary Clearance Route | Recommended Application |
|---|---|---|---|---|
| <6 nm | Rapid extravasation | Deep, diffuse | Renal clearance | Small molecule drugs, not for EPR |
| 10-50 nm | Efficient extravasation via endothelial gaps | High (reaches perivascular region) | Moderate MPS uptake | Optimal for EPR-based tumor accumulation |
| 50-150 nm | Good extravasation | Moderate (peri-vascular) | Significant MPS uptake (liver, spleen) | Common PNP range; balance of load and retention |
| 150-300 nm | Limited extravasation | Low (vascular vicinity) | Rapid MPS sequestration | Macrophage targeting, not optimal for passive EPR |
| >300 nm | Minimal extravasation | Negligible | Rapid MPS clearance | Not suitable for intravenous EPR therapy |
Table 2: Influence of Surface Charge (Zeta Potential) on Nanoparticle Fate
| Zeta Potential Range | Colloidal Stability | Protein Corona (Opsonization) | Cellular Uptake | Impact on EPR |
|---|---|---|---|---|
| Strongly Positive (> +30 mV) | High (electrostatic repulsion) | Heavy, non-specific adsorption | Very high (non-specific, incl. endothelial) | Poor; rapid clearance, potential toxicity |
| Slightly Positive (+5 to +30 mV) | Moderate to High | Moderate, may enhance tumor cell uptake | Enhanced (tumor cell interaction) | Can be beneficial if stability maintained |
| Near-Neutral (-10 to +10 mV) | Low (requires steric stabilizers) | Minimized ("stealth" property) | Low (reduced non-specific uptake) | Optimal for long circulation (EPR prerequisite) |
| Slightly Negative (-10 to -30 mV) | Moderate | Low to moderate | Low to moderate | Good; typical of PEGylated, long-circulating particles |
| Strongly Negative (< -30 mV) | High | High (alternative pathway activation) | Reduced (repulsion by cell membrane) | Suboptimal; may activate complement system |
Table 3: Role of Hydrophobicity in Nanoparticle Performance
| Property | Impact on Drug Loading | Impact on Protein Adsorption | Key Interaction | Design Strategy |
|---|---|---|---|---|
| High Hydrophobicity | High for hydrophobic drugs (e.g., paclitaxel) | Promotes extensive opsonization | Hydrophobic interactions with plasma proteins | Requires shielding (e.g., PEG corona) for EPR |
| Moderate Hydrophobicity | Good for many chemotherapeutics | Moderate; manageable | Can aid in cellular internalization post-extra. | Useful for core-forming polymers (PLGA, PLA) |
| Hydrophilic Surface | Low (unless conjugated) | Minimized ("stealth") | Hydration layer reduces protein adhesion | Critical for achieving long circulation time |
| Amphiphilic Design | High in hydrophobic core | Controlled via hydrophilic shell | Core-shell structure optimal for delivery | Gold standard: Hydrophobic core + PEG shell |
Detailed Experimental Protocols
Protocol 1: Fabrication and Characterization of Size-Varied PNPs (Nanoprecipitation)
- Objective: Synthesize a library of PNPs with controlled diameters from 50-200 nm.
- Materials: Biodegradable polymer (e.g., PLGA), acetone, aqueous surfactant solution (e.g., PVA), magnetic stirrer, probe sonicator, dynamic light scattering (DLS) instrument.
- Method:
- Dissolve PLGA in acetone at a fixed concentration (e.g., 10 mg/mL).
- Prepare an aqueous phase containing 1% w/v PVA.
- Using a syringe pump, add the organic phase dropwise (e.g., 1 mL/min) into the aqueous phase (e.g., 10 mL) under vigorous magnetic stirring (800 rpm).
- To vary size, alter the polymer concentration (5-20 mg/mL) or the aqueous-to-organic phase volume ratio (5:1 to 20:1). Lower polymer concentration and higher aqueous volume yield smaller particles.
- Stir overnight to evaporate acetone.
- Centrifuge (e.g., 21,000 x g, 30 min) and wash pellets 3x with DI water to remove excess surfactant.
- Resuspend in buffer for characterization.
- Measure hydrodynamic diameter and PDI via DLS. Validate with TEM for dry-state size.
Protocol 2: Evaluating the Impact of Surface Charge on Plasma Protein Adsorption
- Objective: Correlate zeta potential with the composition of the protein corona.
- Materials: PNP libraries with varied zeta potential, fetal bovine serum (FBS), centrifuge, SDS-PAGE apparatus, LC-MS/MS facility.
- Method:
- Incubate standardized amounts of PNPs (with positive, neutral, and negative surfaces) in 50% FBS/PBS at 37°C for 1 hour.
- Centrifuge at high speed to pellet the PNPs with their hard protein corona.
- Wash gently with PBS to remove loosely associated proteins.
- Elute proteins from the nanoparticle surface using 2% SDS solution.
- Analyze eluted proteins via SDS-PAGE for a qualitative profile.
- For quantitative analysis, subject samples to trypsin digestion and LC-MS/MS to identify and quantify adsorbed proteins (e.g., albumin, apolipoproteins, immunoglobulins, complement factors).
Protocol 3: In Vivo Validation of EPR Efficacy via Fluorescence Imaging
- Objective: Quantify tumor accumulation of PNPs varying in size and surface charge.
- Materials: Murine tumor xenograft model, near-infrared (NIR) dye (e.g., DiR)-loaded PNPs, IVIS imaging system.
- Method:
- Label PNPs from Protocol 1&2 by incorporating a lipophilic NIR dye into the polymer matrix during formulation.
- Inject dye-loaded PNPs intravenously into tumor-bearing mice (n=5 per group).
- Acquire whole-body fluorescence images at set time points (1, 4, 12, 24, 48 h) post-injection using an IVIS spectrum.
- Euthanize animals at terminal time point (e.g., 48 h), excise tumors and major organs (liver, spleen, kidneys, lungs, heart).
- Image ex vivo organs to quantify fluorescence intensity, correlating to nanoparticle concentration.
- Calculate tumor-to-background ratio (TBR) and % injected dose per gram of tissue (%ID/g). Optimal EPR is indicated by high TBR and %ID/g in tumor relative to liver/spleen.
Signaling and Mechanistic Pathways
Title: PNP Surface Charge Dictates Circulation vs. Clearance Fate
Title: Amphiphilic PNP Structure for Stealth and Loading
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function/Relevance | Example Brands/Types |
|---|---|---|
| Biodegradable Polymers | Forms the nanoparticle matrix; dictates degradation and drug release kinetics. | PLGA, PLA, PCL, Chitosan, Poly(alkyl cyanoacrylates) |
| PEG Derivatives (PEGylation Agents) | Provides hydrophilic stealth corona to reduce opsonization and prolong circulation. | mPEG-PLA, PEG-PLGA diblock copolymers, heterobifunctional PEG (e.g., NHS-PEG-MAL) for ligand conjugation. |
| Dynamic Light Scattering (DLS) Instrument | Measures hydrodynamic particle size, size distribution (PDI), and zeta potential. | Malvern Zetasizer, Brookhaven Instruments. |
| Dialysis Membranes/Tangential Flow Filtration (TFF) | Purifies nanoparticle suspensions, removes organic solvents, and exchanges buffers. | Spectra/Por dialysis tubing, Repligen TFF systems. |
| Near-Infrared (NIR) Fluorescent Dyes | Labels nanoparticles for non-invasive in vivo tracking and biodistribution studies. | DiR, DiD, Cy7, IRDye 800CW. |
| In Vivo Imaging System (IVIS) | Enables real-time, quantitative longitudinal imaging of fluorescently labeled nanoparticle biodistribution. | PerkinElmer IVIS Spectrum, Bruker In-Vivo Xtreme. |
| Model Hydrophobic Drug | Standard compound for testing loading capacity and release profiles. | Paclitaxel, Doxorubicin (base), Curcumin. |
| Differential Scanning Calorimetry (DSC) | Analyzes crystallinity, polymer-drug compatibility, and glass transition temperature (Tg). | TA Instruments DSC, Mettler Toledo DSC. |
Within the landscape of nanomedicine and drug delivery, polymeric nanoparticles (PNPs) stand as a cornerstone technology, particularly for exploiting the Enhanced Permeability and Retention (EPR) effect. The EPR effect, a phenomenon where macromolecules and nanoparticles preferentially accumulate in tumor tissue due to its leaky vasculature and impaired lymphatic drainage, provides a rationale for passive tumor targeting. The composition, type, and innate physicochemical properties of PNPs are critical determinants of their in vivo fate, directly influencing circulation time, biodistribution, tumor accumulation, and ultimate therapeutic efficacy. This guide provides a technical deep-dive into the core aspects of PNPs, framing their design within the strategic context of EPR-mediated delivery.
Core Composition and Synthesis
Polymeric nanoparticles are typically colloidal systems ranging from 10-1000 nm, composed of biodegradable or biocompatible polymers. Their core composition dictates degradation kinetics, drug release profiles, and biocompatibility.
Key Components:
- Polymer Matrix: The primary structural component (e.g., PLGA, chitosan).
- Active Pharmaceutical Ingredient (API): Encapsulated therapeutic (hydrophobic/hydrophilic drugs, nucleic acids, proteins).
- Surfactants/Stabilizers: (e.g., polyvinyl alcohol (PVA), polysorbate 80) used during emulsification to control particle size and prevent aggregation.
- Functional Modifiers: Surface-attached molecules (e.g., PEG for stealth, targeting ligands like antibodies or peptides).
Primary Synthesis Methods:
- Emulsification-Solvent Evaporation: The polymer is dissolved in a volatile organic solvent (e.g., dichloromethane), emulsified in an aqueous phase containing a stabilizer, and the solvent is evaporated to form solid nanoparticles. Ideal for hydrophobic drugs.
- Nanoprecipitation (Solvent Displacement): The polymer and drug are dissolved in a water-miscible solvent (e.g., acetone) and added dropwise to an aqueous phase under stirring. Rapid solvent diffusion yields nanoparticles.
- Ionic Gelation (for polysaccharides): Used for chitosan nanoparticles. The cationic chitosan solution is added to an anionic cross-linker (e.g., tripolyphosphate, TPP), leading to gelation via electrostatic interaction.
Major Types of Polymeric Nanoparticles
PLGA (Poly(lactic-co-glycolic acid))
A synthetic copolymer of lactic acid and glycolic acid, it is FDA-approved for numerous drug delivery applications.
- Innate Properties: Biodegradable, biocompatible. Degradation rate and drug release kinetics can be tuned by altering the LA:GA ratio (higher GA content degrades faster). Hydrophobic, forming a dense matrix.
- EPR Relevance: Unmodified PLGA nanoparticles are often rapidly opsonized and cleared by the mononuclear phagocyte system (MPS), limiting their circulation time and passive accumulation via EPR.
PEG-PLGA (PEGylated PLGA)
PLGA nanoparticles with a surface coating of polyethylene glycol (PEG), either via block copolymer (PLGA-PEG-PLGA) or adsorption.
- Innate Properties: PEG creates a hydrophilic "steric shield" that reduces protein adsorption (opsonization). This "stealth" character prolongs systemic circulation.
- EPR Relevance: The prolonged circulation half-life is essential for maximizing passive tumor targeting via the EPR effect, allowing more nanoparticles to extravasate through tumor vasculature.
Chitosan
A natural, cationic polysaccharide derived from chitin.
- Innate Properties: Biodegradable, biocompatible, mucoadhesive, and can transiently open tight junctions (permeation-enhancing). Its positive charge enables complexation with nucleic acids (siRNA, pDNA) and anionic polymers.
- EPR Relevance: Its positive charge can lead to non-specific interactions with serum components and cells, potentially shortening circulation time. Surface modification (e.g., PEGylation) is often employed to improve pharmacokinetics for EPR.
Other Notable Types
- Poly(ε-caprolactone) (PCL): Slower degrading, more hydrophobic than PLGA.
- Poly(alkyl cyanoacrylates) (PACA): Polymerize in situ, used for rapid drug release.
- Dendrimers: Highly branched, monodisperse polymers with multivalent surfaces.
Table 1: Comparative Properties of Major Polymeric Nanoparticle Types
| Polymer Type | Core Charge | Degradation Timeframe | Key Innate Properties | Primary Synthesis Method | Typical Size Range (nm) |
|---|---|---|---|---|---|
| PLGA | Negative / Neutral | Weeks to Months | Tunable degradation, excellent biocompatibility | Emulsification, Nanoprecipitation | 100-300 |
| PEG-PLGA | Near-Neutral | Weeks to Months | Stealth (reduced opsonization), prolonged circulation | Emulsification, Nanoprecipitation | 80-250 |
| Chitosan | Positive | Hours to Days | Mucoadhesive, permeation-enhancing, bioadhesive | Ionic Gelation, Complex Coacervation | 80-500 |
| PCL | Neutral | Months to Years | Slow degradation, high drug permeability | Emulsification, Nanoprecipitation | 100-400 |
Innate Properties Governing EPR Efficacy
The effectiveness of the EPR effect is not guaranteed; it depends heavily on nanoparticle design.
- Size: Optimal size for EPR is generally considered 10-200 nm. Particles <10 nm undergo renal clearance; >200 nm are filtered by the spleen or may not extravasate efficiently.
- Surface Charge (Zeta Potential): Near-neutral or slightly negative surfaces (e.g., PEG-PLGA) minimize non-specific cellular interactions and MPS uptake, promoting longer circulation. Highly positive or negative surfaces promote rapid clearance.
- Hydrophobicity: Hydrophobic surfaces attract opsonins. Surface hydrophilic modification (PEGylation) is a standard strategy to confer "stealth."
- Drug Loading & Release Profile: High Encapsulation Efficiency (EE) and controlled, sustained release at the target site are crucial. A burst release in circulation can cause systemic toxicity.
Table 2: Impact of Physicochemical Properties on Biological Fate
| Property | Optimal Range for EPR | Consequence of Deviation |
|---|---|---|
| Size | 10 - 200 nm | <10 nm: Renal clearance. >200 nm: Splenic filtration, poor extravasation. |
| Zeta Potential | -10 mV to +10 mV (in plasma) | Strongly positive/negative: Rapid MPS clearance, serum instability. |
| Surface Hydrophilicity | High (Stealth) | High hydrophobicity: Opsonization and rapid clearance by liver/spleen. |
| Stability | >24 hrs in serum | Aggregation leads to size increase and embolization. |
Experimental Protocols
Protocol: Formulation of PLGA Nanoparticles via Emulsification-Solvent Evaporation
Objective: To prepare drug-loaded PLGA nanoparticles. Materials:
- PLGA (50:50, acid-terminated)
- Dichloromethane (DCM)
- Polyvinyl Alcohol (PVA, 1% w/v aqueous solution)
- Drug (e.g., Doxorubicin base)
- Probe sonicator
- Magnetic stirrer
- Rotary evaporator
Procedure:
- Dissolve 100 mg PLGA and 5 mg drug in 5 mL DCM (organic phase).
- Pour the organic phase into 20 mL of 1% PVA solution under probe sonication (70% amplitude, 2 min, on ice).
- Transfer the coarse emulsion to 50 mL of 0.1% PVA solution and stir magnetically for 4 hours to evaporate DCM.
- Centrifuge the nanoparticle suspension at 20,000 rpm for 30 min at 4°C. Wash pellet 3x with distilled water to remove residual PVA and free drug.
- Resuspend the final nanoparticle pellet in 5 mL water or buffer and lyophilize for storage.
Protocol: Evaluation ofIn VitroDrug Release
Objective: To characterize the release kinetics of encapsulated drug. Materials:
- Drug-loaded nanoparticles
- Phosphate Buffered Saline (PBS, pH 7.4) +/- 0.1% Tween 80 (sink condition)
- Dialysis bag (MWCO 12-14 kDa) or Franz diffusion cell
- UV-Vis Spectrophotometer/HPLC
Procedure:
- Place 5 mg of nanoparticles (in 1 mL PBS) into a dialysis bag, sealed at both ends.
- Immerse the bag in 50 mL of release medium (PBS + 0.1% Tween) at 37°C with gentle agitation.
- At predetermined intervals (0.5, 1, 2, 4, 8, 24, 48, 72h...), withdraw 1 mL of the external medium and replace with fresh pre-warmed medium.
- Analyze the drug concentration in the samples using a validated analytical method (e.g., HPLC).
- Calculate cumulative drug release (%) vs. time.
Visualization of Key Concepts
Diagram 1: In vivo fate of polymeric nanoparticles.
Diagram 2: PLGA NP synthesis by emulsification.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Polymeric Nanoparticle Research
| Item / Reagent | Function & Purpose | Key Considerations |
|---|---|---|
| PLGA (50:50, ester-terminated) | Core biodegradable polymer matrix. Standard for controlled release. | LA:GA ratio, molecular weight, and end-group (acid vs. ester) affect degradation rate. |
| Methoxy-PEG-PLGA Copolymer | Forms stealth nanoparticle core with built-in PEG corona. | PEG molecular weight (2k-5k Da) impacts stealth and final particle size. |
| Chitosan (low/medium MW) | Natural cationic polymer for gene/drug delivery. | Degree of deacetylation (>75%) and molecular weight determine solubility & bioactivity. |
| Polyvinyl Alcohol (PVA, 87-89% hydrolyzed) | Surfactant/Stabilizer in emulsion methods. | Hydrolysis degree affects residual acetate groups, influencing stability and nanoparticle surface. |
| Dialysis Tubing (MWCO 12-14 kDa) | Purification and in vitro drug release studies. | Must be pretreated to remove glycerin. MWCO should be 5-10x smaller than nanoparticle size. |
| Dynamic Light Scattering (DLS) / Zetasizer | Instrument for measuring particle size (hydrodynamic diameter), PDI, and zeta potential. | Sample must be dilute and free of dust. Zeta potential measured in low conductivity buffer. |
| Tripolyphosphate (TPP) | Ionic cross-linker for forming chitosan nanoparticles via ionic gelation. | Concentration and chitosan:TPP volume ratio critically control particle size and stability. |
| Cyanoacrylate Superglue | Quick-sealing dialysis bags or device assembly. | Ensure it is fully cured before immersion to avoid contaminating the release medium. |
Why Polymers? Advantages for EPR-Mediated Delivery Over Other Nanomaterials
This whitepaper is situated within a comprehensive thesis investigating the Enhanced Permeability and Retention (EPR) effect as a cornerstone of solid-tumor targeting. The central premise is that while the EPR effect provides a passive targeting paradigm for nanomaterials, its successful exploitation is critically dependent on the physicochemical and biological properties of the carrier. This document argues that polymeric nanoparticles (PNPs) offer a superior and more versatile platform for capitalizing on the EPR mechanism compared to other nanomaterial classes, such as liposomes, inorganic nanoparticles, and dendrimers. Their advantages stem from unparalleled tunability in composition, architecture, and surface functionality, which directly translates to enhanced control over pharmacokinetics, biodistribution, tumor accumulation, and drug release kinetics.
Comparative Advantages of Polymeric Nanoparticles
The superiority of PNPs in EPR-mediated delivery is multi-faceted, grounded in material science and pharmacokinetic principles.
- Material and Structural Tunability: Polymers offer a vast chemical space (poly(lactic-co-glycolic acid) (PLGA), poly(ε-caprolactone) (PCL), chitosan, poly(alkyl cyanoacrylates), etc.). Their molecular weight, copolymer ratio, and block architecture can be precisely engineered to modulate degradation rates from hours to months, aligning drug release with therapeutic needs.
- High Drug Loading and Versatility: PNPs can encapsulate hydrophobic drugs within their core, conjugate drugs covalently to the backbone, or electrostatically complex with biomacromolecules (e.g., siRNA, pDNA). This enables the delivery of a wider range of payloads compared to some constrained systems.
- Stability and Shelf-Life: Solid polymeric matrices generally offer greater in vivo and storage stability than lipid-based systems (e.g., liposomes), reducing premature drug leakage and extending shelf-life.
- Surface Engineering Facilitation: The polymer terminus or backbone can be readily functionalized with targeting ligands, PEG for stealth, or environmentally responsive moieties (e.g., pH-sensitive linkers) to create "smart" systems that respond to the tumor microenvironment.
Table 1: Quantitative Comparison of Nanomaterial Platforms for EPR-Mediated Delivery
| Property | Polymeric Nanoparticles (e.g., PLGA) | Liposomes | Inorganic NPs (e.g., Mesoporous Silica) | Dendrimers |
|---|---|---|---|---|
| Typical Size Range (nm) | 20-500 | 50-350 | 20-200 | 2-10 (core), >20 with surface mod. |
| Drug Loading Capacity (%) | 5-50 (High) | 1-10 (Low-Mod) | 10-40 (High) | 10-35 (High) |
| Release Profile Control | Excellent (days to months via polymer degradation) | Moderate (burst release common) | Good (pore gating possible) | Good (surface-controlled) |
| In Vivo Stability | High | Low-Moderate (fusion, leakage) | Very High | High |
| Scalability & Cost | Good, moderate cost | Excellent, established manufacture | Moderate, cost varies | Challenging, high cost |
| Functionalization Ease | Excellent | Good | Excellent | Excellent (monodisperse) |
| Clearance Pathway | Biodegradable polymers: metabolic clearance | Enzymatic degradation, RES uptake | Often non-biodegradable; long-term safety concerns | Renal clearance (size-dependent) |
Key Experimental Protocols in PNP Research for EPR
Protocol 1: Nanoprecipitation for PNP Formulation Objective: To fabricate biodegradable PNPs (e.g., PLGA) encapsulating a hydrophobic drug. Materials: PLGA polymer, hydrophobic drug (e.g., paclitaxel), acetone or acetonitrile (organic phase), aqueous solution containing a stabilizer (e.g., 0.5% w/v polyvinyl alcohol, PVA). Method:
- Dissolve PLGA and the drug at a desired ratio (e.g., 10:1 w/w) in the organic solvent.
- Using a syringe pump or pipette, rapidly inject the organic solution into the vigorously stirring aqueous phase (typical organic-to-aqueous ratio of 1:5 to 1:10).
- Stir the mixture for 3-6 hours at room temperature to allow for complete organic solvent evaporation and nanoparticle hardening.
- Centrifuge the suspension (e.g., 20,000 x g, 30 min) to pellet NPs. Wash with water to remove excess stabilizer.
- Resuspend the NP pellet in buffer or lyophilize for storage. Characterize for size (DLS), polydispersity (PDI), zeta potential, and drug loading (HPLC).
Protocol 2: In Vivo Evaluation of EPR-Mediated Tumor Accumulation Objective: To quantitatively compare the tumor accumulation of polymeric NPs vs. other nanocarriers. Materials: Fluorescently or radiolabeled (e.g., with Cy5.5 or ¹¹¹In) PNPs and control NPs, mouse model with subcutaneous tumor (e.g., 4T1 breast carcinoma, ~300 mm³). Method:
- Administer a standardized dose (e.g., 5 mg/kg nanoparticle equivalent) via tail vein injection to tumor-bearing mice (n=5 per group).
- At predetermined time points (e.g., 1, 4, 24, 48 h), euthanize animals and collect blood, major organs (liver, spleen, kidneys, heart, lungs), and tumor.
- Homogenize tissues and quantify fluorescence/radioactivity using an IVIS imaging system or gamma counter, respectively.
- Calculate % injected dose per gram of tissue (%ID/g). Key metrics: Tumor AUC (Area Under the Curve), Tumor-to-Muscle ratio, and Tumor-to-Liver ratio (indicative of stealth properties).
Visualization of Key Concepts
Title: The EPR Pathway for Polymeric Nanoparticles
Title: PNP Development and Evaluation Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Research Reagents for PNP Development Targeting the EPR Effect
| Item | Function & Rationale |
|---|---|
| Biodegradable Polymers (PLGA, PCL) | The core matrix material. PLGA's degradation rate and release profile are tuned by its LA:GA ratio. Provides controlled release and biocompatibility. |
| DSPE-mPEG (Lipid-PEG) | A common stealth coating agent. Inserted during formulation to create a hydrophilic, sterically stabilizing corona that reduces opsonization and extends circulation half-life. |
| Polyvinyl Alcohol (PVA) | A common stabilizer/surfactant used in emulsion-based NP formulation (e.g., single/double emulsion). Controls particle size and prevents aggregation during synthesis. |
| Cyanine Dyes (Cy5.5, DiR) | Near-infrared fluorescent labels for in vivo and ex vivo imaging. Allows non-invasive tracking of biodistribution and tumor accumulation over time. |
| Cell-Penetrating Peptides (e.g., TAT) | Ligands conjugated to NP surface to enhance intracellular delivery post-extravasation, moving beyond passive EPR to active targeting. |
| pH-Sensitive Linkers (e.g., Hydrazone) | Used to conjugate drugs to the polymer. Stable in blood (pH 7.4) but cleave in the acidic tumor microenvironment or endo/lysosomes (pH 5-6), enabling triggered release. |
| Size Exclusion Chromatography (SEC) Media | For purification of functionalized polymers or NPs to remove unreacted precursors, ensuring batch consistency and accurate characterization. |
| Dynamic Light Scattering (DLS) & Zeta Potential Analyzer | Essential instrument for measuring hydrodynamic diameter, polydispersity index (PDI), and surface charge (zeta potential) – critical quality attributes for EPR. |
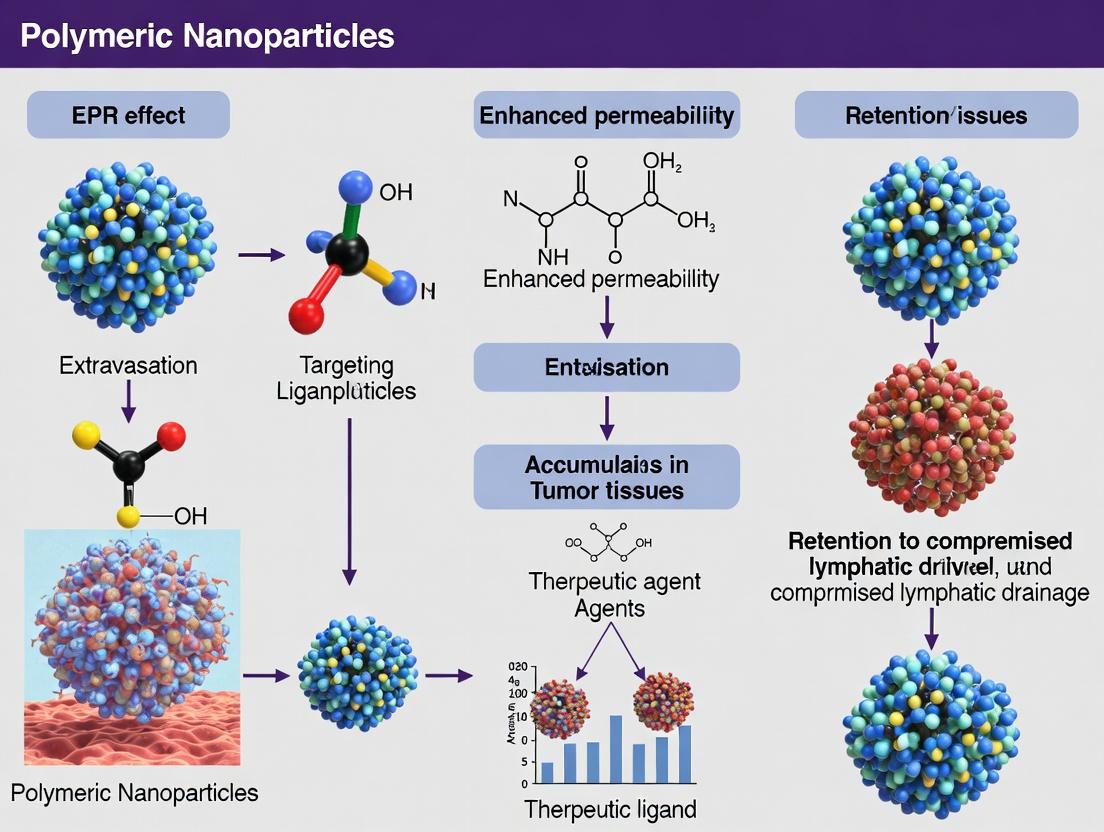
The Enhanced Permeability and Retention (EPR) effect, first described by Matsumura and Maeda, remains a foundational concept in tumor-targeted drug delivery, particularly for nanomedicines like polymeric nanoparticles. This whitepaper, framed within a broader thesis on advancing EPR-based therapeutics, examines the critical and often underappreciated heterogeneity of the EPR effect. Its magnitude and consistency are not universal but vary significantly across tumor types, anatomical locations, and between individuals. Understanding this variability is paramount for researchers and drug development professionals to design effective nanocarriers, select appropriate preclinical models, and stratify patients in clinical trials.
Quantitative Analysis of EPR Heterogeneity
The variability of the EPR effect can be quantified through key parameters: vascular permeability (often measured as the permeability-surface area product, PS), pore cutoff size, interstitial fluid pressure (IFP), and nanoparticle accumulation (%ID/g – percentage of injected dose per gram of tissue). The following tables consolidate recent experimental data.
Table 1: Variability Across Tumor Types & Models
| Tumor Model / Type | Vascular Permeability (PS, µL/min/g) | Pore Cutoff Size (nm) | IFP (mmHg) | NP Accumulation (%ID/g) | Key Reference |
|---|---|---|---|---|---|
| Murine C26 Colon Carcinoma | 45.2 ± 12.1 | 100-200 | 12.3 ± 3.1 | 8.5 ± 2.1 | [1] |
| Murine B16F10 Melanoma | 22.7 ± 8.5 | 50-100 | 18.5 ± 4.2 | 4.2 ± 1.5 | [1] |
| Murine 4T1 Breast Carcinoma | 67.8 ± 15.3 | 150-300 | 8.7 ± 2.8 | 10.3 ± 3.0 | [2] |
| Patient-Derived Xenograft (PDX) - Pancreatic | 15.4 ± 6.2 | 30-80 | 32.1 ± 7.5 | 1.8 ± 0.9 | [3] |
| Patient-Derived Xenograft (PDX) - Soft Tissue Sarcoma | 28.9 ± 9.8 | 80-150 | 24.5 ± 5.5 | 3.5 ± 1.2 | [3] |
Table 2: Impact of Tumor Location (Orthotopic vs. Subcutaneous)
| Tumor Cell Line | Implantation Site | NP Accumulation (%ID/g) | Vessel Density (vessels/mm²) | Notes |
|---|---|---|---|---|
| Glioblastoma (U87) | Intracranial (Orthotopic) | 1.2 ± 0.5 | 450 ± 120 | Blood-brain barrier influence |
| Glioblastoma (U87) | Subcutaneous | 5.5 ± 1.8 | 280 ± 85 | More permeable, non-CNS environment |
| Pancreatic (Panc02) | Pancreatic (Orthotopic) | 2.1 ± 0.7 | 150 ± 50 | High stromal density, high IFP |
| Pancreatic (Panc02) | Subcutaneous | 6.8 ± 2.1 | 220 ± 70 | Less stroma, more typical EPR |
Experimental Protocols for Assessing EPR
Protocol:In VivoQuantification of Nanoparticle Tumor Accumulation
Objective: To measure the biodistribution and tumor accumulation of fluorescently labeled or radiolabeled polymeric nanoparticles.
- Nanoparticle Preparation: Synthesize polymeric nanoparticles (e.g., PLGA-PEG) and label with a near-infrared dye (e.g., DiR, Cy7.5) or a radioisotope (e.g., ^111In, ^64Cu).
- Tumor Models: Establish heterogeneous tumor models (subcutaneous, orthotopic, PDX models of varying histology).
- Administration: Inject nanoparticles intravenously via the tail vein in mice (dose: 5-20 mg/kg, 100-200 µL volume) when tumors reach 200-500 mm³.
- In Vivo Imaging: For fluorescent probes, use an IVIS Spectrum or similar system at multiple time points (1, 4, 24, 48 h). Acquire fluorescence and photographic images. Draw regions of interest (ROI) over tumor and major organs.
- Ex Vivo Quantification: At terminal time points (e.g., 24 h and 48 h), sacrifice animals. Excise tumors, liver, spleen, kidneys, lungs, heart, and a blood sample. Weigh tissues.
- Fluorescence: Homogenize tissues, extract dye in appropriate solvent, measure fluorescence with a plate reader against a standard curve.
- Radiolabel: Count tissue radioactivity with a gamma counter.
- Data Analysis: Calculate %ID/g = (Radioactivity or fluorescence in tissue / Total injected dose) / Tissue weight * 100%. Compare across tumor models and locations.
Protocol: Multiplex Immunofluorescence Analysis of Tumor Vasculature
Objective: To characterize tumor vessel density, pericyte coverage, and endothelial fenestration as determinants of EPR heterogeneity.
- Tissue Collection: Flash-freeze tumor tissues in optimal cutting temperature (OCT) compound or fix in 4% paraformaldehyde and paraffin-embed.
- Sectioning: Cut 5-10 µm thick sections using a cryostat or microtome.
- Staining: Perform multiplex immunofluorescence staining.
- Blocking: Incubate with 5% normal serum/1% BSA for 1 h.
- Primary Antibodies (cocktail): Anti-CD31 (endothelial cells, 1:100), Anti-α-SMA (pericytes, 1:200), Anti-Laminin (basement membrane, 1:100), Anti-Collagen IV (basement membrane, 1:100). Incubate overnight at 4°C.
- Secondary Antibodies: Use species-specific secondary antibodies conjugated to distinct fluorophores (e.g., Alexa Fluor 488, 555, 647). Incubate for 1 h at RT. Include DAPI for nuclei.
- Imaging: Acquire high-resolution images using a confocal or multiphoton microscope. Capture multiple fields per tumor (≥5).
- Image Analysis: Use software (e.g., ImageJ, HALO, Imaris) to:
- Calculate microvessel density (MVD): CD31+ structures/mm².
- Determine pericyte coverage index: (% of CD31+ vessel length co-localized with α-SMA).
- Assess basement membrane continuity.
Visualizing Key Concepts and Pathways
Diagram 1: Factors Governing EPR Heterogeneity (79 chars)
Diagram 2: Protocol for EPR Heterogeneity Study (85 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for EPR Heterogeneity Research
| Item / Reagent | Function / Application in EPR Studies | Example Vendor / Catalog (Representative) |
|---|---|---|
| Fluorescent/Radiometric Polymeric NPs | Core tool for tracking tumor accumulation; PLGA-PEG, HPMA copolymers are common. Must be well-characterized (size, zeta potential, PDI). | Avanti Polar Lipids (nanoparticle kits), Sigma-Aldrich (PLGA resins), custom synthesis. |
| Near-Infrared Dyes (Cy7, DiR, IRDye) | For non-invasive in vivo imaging (IVIS) and ex vivo tissue quantification of nanoparticle biodistribution. | Lumiprobe, LI-COR, Thermo Fisher Scientific. |
| Matrigel Basement Membrane Matrix | For consistent implantation of certain tumor cells, especially in subcutaneous models; can influence early vascularization. | Corning, 356234. |
| Primary Antibodies for mIF | Critical for characterizing tumor vasculature and TME: Anti-CD31 (vascular density), Anti-α-SMA (pericytes), Anti-Collagen IV (basement membrane). | Abcam, Cell Signaling Technology, BioLegend. |
| IVIS Spectrum Imaging System | Primary instrument for longitudinal, non-invasive quantification of fluorescent nanoparticle signal in tumors and organs. | PerkinElmer. |
| Laser Doppler Perfusion Imager | Measures real-time blood flow in superficial tumors; correlates with vascular functionality and potential EPR. | Moor Instruments, Perimed. |
| Wingate Catheter & Pressure Transducer | For direct measurement of Interstitial Fluid Pressure (IFP), a major barrier to nanoparticle extravasation. | Millar, Inc. (SPR-1000). |
| Patient-Derived Xenograft (PDX) Models | Provides clinically relevant tumor stroma and vascular phenotypes, essential for translational EPR studies. | The Jackson Laboratory, Champions Oncology, Charles River. |
| Image Analysis Software (HALO, Imaris) | For quantitative analysis of multiplex immunofluorescence (mIF) images to derive vessel metrics and spatial relationships. | Indica Labs, Oxford Instruments. |
Designing EPR-Compliant Carriers: Synthesis, Functionalization, and Payload Strategies
Within the landscape of polymeric nanoparticle research for drug delivery, synthesis methodology is a primary determinant of nanoparticle characteristics and, consequently, in vivo fate via the Enhanced Permeation and Retention (EPR) effect. This technical guide details three core synthesis techniques—emulsion, nanoprecipitation, and polymerization methods—framed within the context of optimizing nanoparticles for passive tumor targeting.
Emulsion-Based Synthesis
Emulsion techniques involve the dispersion of a polymer-containing organic phase into an aqueous continuous phase, stabilized by surfactants.
Key Experimental Protocol: Single Emulsion (Oil-in-Water)
- Dissolution: Dissolve 100-500 mg of biodegradable polymer (e.g., PLGA) and the hydrophobic active compound in 5-20 mL of a water-immiscible organic solvent (e.g., dichloromethane or ethyl acetate).
- Emulsification: Add the organic phase dropwise to 50-200 mL of an aqueous surfactant solution (e.g., 1-5% w/v polyvinyl alcohol, PVA) under constant stirring (500-1000 rpm) to form a coarse emulsion.
- Homogenization: Subject the coarse emulsion to high-energy homogenization (e.g., probe sonication at 100 W for 60-120 seconds or high-pressure homogenization at 10,000-15,000 psi for 3-5 cycles) to form a fine oil-in-water (O/W) nanoemulsion.
- Solvent Removal: Stir the nanoemulsion for 3-12 hours to allow for organic solvent evaporation, or employ reduced pressure. This hardens the polymer droplets into solid nanoparticles.
- Purification: Centrifuge the suspension (e.g., 20,000-25,000 x g, 30-60 minutes), discard the supernatant containing free surfactant and unencapsulated drug, and resuspend the pellet in deionized water or buffer. Repeat 2-3 times.
- Lyophilization: For storage, freeze-dry the purified nanoparticle suspension, often with a cryoprotectant (e.g., 5% w/v trehalose).
Research Reagent Solutions
| Reagent/Material | Function in Synthesis |
|---|---|
| Poly(lactic-co-glycolic acid) (PLGA) | Biodegradable copolymer forming the nanoparticle matrix; degradation rate tunable by LA:GA ratio. |
| Polyvinyl Alcohol (PVA) | Surfactant that stabilizes the oil-water interface during emulsification, preventing coalescence. |
| Dichloromethane (DCM) | Volatile, water-immiscible organic solvent that dissolves hydrophobic polymers and drugs. |
| Trehalose | Cryoprotectant that prevents nanoparticle aggregation and protects structure during freeze-drying. |
Title: Single Emulsion (O/W) Nanoparticle Synthesis Workflow
Nanoprecipitation (Solvent Displacement)
This method relies on the interfacial deposition of polymer following the displacement of a water-miscible solvent from a organic phase into an aqueous phase.
Key Experimental Protocol
- Organic Phase Preparation: Dissolve 10-100 mg of polymer and the active compound in 5-20 mL of a water-miscible solvent (e.g., acetone or tetrahydrofuran).
- Aqueous Phase Preparation: Prepare 50-200 mL of deionized water or an aqueous solution with a stabilizing agent (e.g., 0.1-1% w/v poloxamer).
- Precipitation: Under moderate magnetic stirring (300-600 rpm), inject the organic phase rapidly (e.g., via syringe pump) into the aqueous phase.
- Formation & Stabilization: The immediate diffusion of the organic solvent into water causes a decrease in interfacial polarity, leading to the spontaneous precipitation of polymer into nanoparticles. The stabilizer adsorbs to nascent particles.
- Solvent Removal: Stir for 1-3 hours to allow for complete diffusion and evaporation of the organic solvent.
- Concentration & Purification: Use rotary evaporation or ultrafiltration to concentrate and wash the nanoparticle suspension. Lyophilize as needed.
Polymerization Methods
This involves the in situ synthesis of the polymer matrix via monomer reaction. Emulsion polymerization is most common for drug delivery nanoparticles.
Key Experimental Protocol: Emulsion Polymerization
- Aqueous Phase Prep: Charge a reactor with 100-500 mL of degassed deionized water and surfactant (e.g., sodium dodecyl sulfate).
- Monomer Addition: Add the hydrophobic monomer (e.g., alkyl cyanoacrylates, methyl methacrylate) and drug, typically 1-10% of aqueous phase volume, under inert atmosphere (N₂).
- Initiation & Polymerization: Heat the mixture to reaction temperature (e.g., 70-80°C). Add a water-soluble initiator (e.g., potassium persulfate, KPS). Polymerization proceeds within monomer-swollen micelles.
- Reaction Completion: Stir for 2-8 hours until monomer conversion is complete.
- Cooling & Purification: Cool to room temperature. Purify via extensive dialysis or tangential flow filtration to remove surfactant, unreacted monomer, and initiator residues.
Quantitative Comparison of Core Synthesis Techniques
Table 1: Key Characteristics and Output Parameters of Core Synthesis Techniques
| Parameter | Emulsion (Single O/W) | Nanoprecipitation | Emulsion Polymerization |
|---|---|---|---|
| Typical Polymer | Pre-formed (PLGA, PLA) | Pre-formed (PLGA, PLA, PCL) | Synthesized in situ (PACA, PMMA) |
| Organic Solvent | Water-immiscible (DCM, EA) | Water-miscible (Acetone, THF) | Monomer (often acts as solvent) |
| Energy Input | High (Homogenization) | Low (Spontaneous) | Moderate (Thermal/Chemical) |
| Typical Size Range | 100 - 500 nm | 50 - 250 nm | 50 - 300 nm |
| Drug Loading | Moderate to High | Low to Moderate | Variable (often lower) |
| Scalability | Excellent (HPH scalable) | Moderate (mixing dynamics critical) | Excellent (industrial precedent) |
| Residual Concerns | Surfactant, Solvent | Solvent | Monomer, Initiator, Surfactant |
Title: Nanoparticle Properties Dictating EPR Effect Outcomes
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Polymeric Nanoparticle Synthesis and Characterization
| Category | Item | Function / Purpose |
|---|---|---|
| Polymers | PLGA (50:50, 75:25) | Tunable degradation for controlled release. Gold standard matrix. |
| Polymers | Poly(ε-caprolactone) (PCL) | Slower degrading, more hydrophobic polymer for sustained release. |
| Polymers | Poly(alkyl cyanoacrylate) (PACA) | Polymerizes in situ; offers high drug encapsulation efficiency. |
| Surfactants | Polyvinyl Alcohol (PVA) | Stabilizer in emulsion methods; influences surface properties. |
| Surfactants | Poloxamer 407 (Pluronic F127) | Non-ionic stabilizer for nanoprecipitation; can enhance biocompatibility. |
| Solvents | Dichloromethane (DCM) | Common solvent for hydrophobic polymers in emulsion techniques. |
| Solvents | Acetone | Water-miscible solvent for nanoprecipitation; fast diffusion rate. |
| Characterization | Dynamic Light Scattering (DLS) | Measures hydrodynamic diameter, PDI, and zeta potential. |
| Characterization | Dialysis Tubing (MWCO) | Purifies nanoparticles by removing small molecules (surfactant, solvent). |
| Characterization | Trehalose / Mannitol | Cryoprotectant to prevent aggregation during lyophilization. |
The selection of synthesis technique directly engineers nanoparticle physicochemical properties (size, surface charge, release kinetics), which are non-negotiable prerequisites for leveraging the EPR effect. Emulsion methods offer robust encapsulation, nanoprecipitation excels in simplicity and small size, and polymerization allows for novel matrix design. Mastery of these core techniques enables the rational design of polymeric nanoparticles tailored for optimal passive targeting and therapeutic efficacy in oncology research.
Polyethylene glycol (PEG) conjugation, or PEGylation, is a cornerstone chemical strategy in nanomedicine, critically enabling the enhanced performance of therapeutic nanoparticles (NPs). Its primary roles—extending systemic circulation time and conferring "stealth" properties—are fundamental to leveraging the Enhanced Permeability and Retention (EPR) effect. The EPR effect, a phenomenon wherein macromolecules and nanoparticles preferentially accumulate in tumor tissue due to leaky vasculature and impaired lymphatic drainage, forms the central thesis of many cancer drug delivery platforms. However, the utility of the EPR effect is contingent upon nanoparticles evading the mononuclear phagocyte system (MPS) and persisting in the bloodstream long enough to reach and extravasate at the target site. Unmodified nanoparticles are rapidly opsonized and cleared by the liver and spleen. PEGylation addresses this by creating a hydrophilic, steric barrier on the nanoparticle surface, reducing protein adsorption (opsonization) and subsequent phagocytic recognition. This technical guide explores the mechanisms, methodologies, and quantitative data underpinning PEGylation's role in optimizing polymeric nanoparticles for EPR-mediated delivery.
Mechanisms of Stealth and Pharmacokinetic Enhancement
The stealth effect of PEG is governed by several interrelated mechanisms:
- Steric Repulsion: The flexible, hydrophilic PEG chains create a dense, hydrated "cloud" around the nanoparticle. This physical barrier increases the distance between the nanoparticle surface and approaching opsonins or phagocytic cell membranes, weakening hydrophobic and van der Waals interactions.
- Reduced Opsonization: The hydrated layer minimizes the adsorption of plasma proteins (e.g., immunoglobulins, complement proteins) that tag particles for MPS clearance. The chain mobility and neutrality of PEG further discourage electrostatic interactions with proteins.
- Brush vs. Mushroom Conformation: The conformation of surface-grafted PEG chains, dictated by grafting density (σ) and molecular weight (MW), is critical. At low density, chains adopt a "mushroom" conformation, offering limited protection. At high density, chains extend into a "brush" conformation, providing optimal steric shielding. The scaling relationship is defined by the Flory radius.
Key Signaling Pathway: PEGylated NP Avoidance of MPS Clearance
The following diagram illustrates the molecular and cellular interactions determining the fate of PEGylated versus non-PEGylated nanoparticles.
Diagram Title: PEGylation-Driven MPS Evasion Pathway
Experimental Protocols for Evaluating PEGylation Efficacy
Protocol: Synthesis of PEGylated Polymeric Nanoparticles (e.g., PLGA-PEG)
Objective: To prepare PEG-PLGA copolymer nanoparticles using the nanoprecipitation or emulsion-solvent evaporation method. Materials:
- PEG-PLGA diblock or triblock copolymer (e.g., PLGA-PEG-COOH, MW 15k-5k Da).
- Organic solvent (Acetone or Ethyl Acetate).
- Aqueous phase (Deionized Water or PBS, pH 7.4).
- Probe sonicator or high-pressure homogenizer.
- Magnetic stirrer.
- Rotary evaporator or dialysis tubing (MWCO 12-14 kDa).
Procedure (Nanoprecipitation):
- Dissolve 50 mg of PEG-PLGA copolymer in 5 mL of acetone under mild stirring.
- Using a syringe pump, slowly inject (1 mL/min) the organic solution into 20 mL of rapidly stirring deionized water.
- Stir the mixture overnight at room temperature to allow for complete solvent evaporation and nanoparticle hardening.
- Concentrate the nanoparticle suspension using a rotary evaporator (gentle vacuum) or purify via dialysis against DI water for 4 hours.
- Filter the suspension through a 0.8/0.45 µm membrane filter. Characterize for size (DLS), zeta potential, and PEG surface density.
Protocol: In Vivo Pharmacokinetic (PK) and Biodistribution Study
Objective: To quantify the blood circulation half-life and tissue biodistribution of PEGylated vs. non-PEGylated NPs. Materials:
- Cy7- or DiR-labeled PEGylated and non-PEGylated nanoparticles.
- Animal model (e.g., BALB/c mice with/without tumor xenografts).
- IVIS Spectrum or similar in vivo imaging system.
- Heparinized capillary tubes for blood collection.
- Tissue homogenizer.
Procedure:
- Administer a dose of 5 mg/kg (nanoparticle weight) via tail vein injection to groups of mice (n=5 per formulation).
- At pre-determined time points (e.g., 5 min, 30 min, 2h, 8h, 24h, 48h), collect ~20 µL of blood retro-orbitally. Lyse blood cells and measure fluorescence intensity (FI) using a plate reader.
- Plot blood FI vs. time. Calculate pharmacokinetic parameters (t1/2α, t1/2β, AUC) using non-compartmental analysis.
- At terminal time points (e.g., 24h and 48h), euthanize animals, harvest major organs (heart, liver, spleen, lungs, kidneys, tumor), and image ex vivo using IVIS.
- Quantify fluorescence in each organ, normalize to tissue weight, and express as % injected dose per gram (%ID/g).
Table 1: Impact of PEG MW and Density on Nanoparticle Properties and PK
| PEG Molecular Weight (Da) | Grafting Density (chains/nm²) | Hydrodynamic Size (nm, DLS) | Zeta Potential (mV) | Blood Circulation Half-life (t1/2β, h) | Liver Accumulation (%ID/g at 24h) |
|---|---|---|---|---|---|
| None (Plain PLGA) | 0 | 150 ± 10 | -25.5 ± 1.5 | 0.8 ± 0.2 | 65.2 ± 8.1 |
| 2,000 | 0.5 | 155 ± 8 | -20.1 ± 2.0 | 2.5 ± 0.5 | 45.3 ± 5.7 |
| 2,000 | 1.5 | 165 ± 12 | -15.3 ± 1.8 | 5.1 ± 1.1 | 28.4 ± 4.2 |
| 5,000 | 0.5 | 170 ± 9 | -18.5 ± 2.1 | 8.3 ± 1.8 | 22.1 ± 3.9 |
| 5,000 | 1.5 | 190 ± 15 | -8.2 ± 1.5 | 18.7 ± 3.2 | 12.5 ± 2.8 |
| 10,000 | 0.3 | 200 ± 18 | -12.4 ± 1.7 | 12.5 ± 2.5 | 18.9 ± 3.5 |
Note: Data is representative and synthesized from recent literature. PLGA core assumed. Higher PEG MW and density increase size, reduce zeta magnitude, and dramatically enhance half-life while reducing liver uptake.
Table 2: Tumor Accumulation via EPR: PEGylated vs. Non-PEGylated Formulations
| Nanoparticle Formulation | Circulation t1/2β (h) | Tumor Accumulation (%ID/g at 24h) | Tumor-to-Liver Ratio (24h) | Primary Clearance Organ |
|---|---|---|---|---|
| Plain PLGA NP | 0.8 | 2.1 ± 0.5 | 0.03 | Liver (>65% ID/g) |
| PLGA-PEG2k (Low Density) | 5.1 | 4.5 ± 1.2 | 0.16 | Liver |
| PLGA-PEG5k (Optimal) | 18.7 | 8.9 ± 2.1 | 0.71 | Liver/Kidneys |
| PLGA-PEG10k (High MW) | 12.5 | 6.8 ± 1.8 | 0.36 | Spleen/Kidneys |
Note: Optimal PEGylation (e.g., 5k Da at high density) maximizes the trade-off between long circulation and effective tumor extravasation, leading to the highest tumor-to-liver ratio, a key metric for EPR efficacy.
Experimental Workflow: From Synthesis to In Vivo Validation
Diagram Title: PEGylated NP Development and EPR Validation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for PEGylation and Stealth Nanoparticle Research
| Item | Function/Benefit | Example Vendor/Product |
|---|---|---|
| Functional PEG-Polymers | Diblock/triblock copolymers with end-group chemistry (COOH, NH2, MAL, NHS) for conjugation. Enable controlled NP surface engineering. | Sigma-Aldrich (PEG-PLGA), Nanocs (PEG-PLGA-COOH), Laysan Bio (mPEG-SH). |
| Fluorescent Lipophilic Dyes | For in vivo and cellular tracking of nanoparticles. High stability and minimal dye leakage are critical. | Thermo Fisher (DiD, DiR, DIR-BOA), PromoCell (PKH26, PKH67). |
| Opsonin & Complement Assay Kits | Quantify protein corona composition (e.g., C3, IgG, albumin) on NPs to directly measure stealth effect. | Hycult Biotech (Human C3a ELISA), Abcam (Complement C3 ELISA). |
| Dynamic Light Scattering (DLS) & Zeta Potential Analyzer | Measures hydrodynamic diameter, PDI, and surface charge—critical for characterizing PEG layer and colloidal stability. | Malvern Panalytical (Zetasizer Nano ZS), Horiba (SZ-100). |
| MicroBCA or Coomassie Plus Protein Assay | Quantifies total protein adsorbed onto nanoparticles after incubation in plasma/serum (protein corona study). | Thermo Fisher (MicroBCA Protein Assay Kit). |
| Dialysis Membranes (Float-A-Lyzer) | For purifying and buffer-exchanging nanoparticle suspensions; MWCO selection is crucial to retain NPs while removing impurities. | Spectrum Labs (Float-A-Lyzer G2, MWCO 100kDa). |
| In Vivo Imaging System (IVIS) | Enables non-invasive, longitudinal tracking of fluorescently labeled nanoparticles in live animals for PK/BD studies. | PerkinElmer (IVIS Spectrum), Bruker (In-Vivo Xtreme). |
PEGylation remains the gold standard for engineering stealth in polymeric nanoparticles aimed at exploiting the EPR effect. The quantitative data and protocols outlined herein provide a framework for optimizing PEG parameters (MW, density, conformation) to maximize circulation time and tumor accumulation. However, challenges such as potential immunogenicity (anti-PEG antibodies) and the "PEG dilemma"—where excessive shielding can hinder target cell uptake—drive ongoing research. Emerging strategies include the development of cleavable PEG linkages, zwitterionic polymers, and dynamic surface coatings. A deep understanding of PEGylation's role, as detailed in this guide, is fundamental for researchers designing the next generation of EPR-optimized nanotherapeutics.
Within the evolving thesis on enhancing the therapeutic index of nanomedicines, the exploitation of the Enhanced Permeability and Retention (EPR) effect has been a cornerstone. While passive targeting via the EPR effect provides a foundational tumor accumulation strategy, its heterogeneity and limited cellular internalization present significant limitations. This guide details the technical integration of active, ligand-mediated targeting with the passive EPR framework, creating a synergistic delivery system for improved specificity and efficacy in cancer therapy.
Theoretical Foundation and Synergistic Rationale
The EPR effect, driven by leaky tumor vasculature and impaired lymphatic drainage, facilitates the passive accumulation of nanoparticles (typically 20-200 nm) in the tumor interstitium. However, this accumulation is often non-uniform and confined to perivascular regions. Ligand-mediated active targeting involves surface-functionalizing nanoparticles with moieties (e.g., folic acid, RGD peptides, transferrin) that bind to receptors overexpressed on cancer cells.
Synergy is achieved sequentially:
- Passive Phase: Polymeric nanoparticles (e.g., PLGA, chitosan, PEG-PLGA copolymers) utilize the EPR effect for primary tumor enrichment.
- Active Phase: Surface-conjugated ligands bind specifically to cell-surface receptors, triggering receptor-mediated endocytosis. This enhances cellular uptake, overcomes multidrug resistance efflux pumps, and can facilitate transcytosis for deeper tumor penetration.
The combination results in a multiplicative effect: higher intratumoral concentration (EPR) coupled with more efficient cell-specific internalization (ligand).
Diagram Title: Sequential Synergy of EPR and Ligand Targeting
Table 1: In Vivo Performance of Passive vs. Active Targeted Nanoparticles
| Nanoparticle Formulation (Polymer-Ligand) | Tumor Model | % Injected Dose/g (Passive) | % Injected Dose/g (Active) | Relative Uptake Increase | Ref. Year |
|---|---|---|---|---|---|
| PEG-PLGA (Non-targeted) | Murine 4T1 Breast | 3.8 ± 0.5 | - | - | 2023 |
| PEG-PLGA-Folate | Murine 4T1 Breast | - | 8.2 ± 0.9 | 2.2x | 2023 |
| PLGA-PEG (Non-targeted) | Murine U87MG Glioblastoma | 2.1 ± 0.3 | - | - | 2024 |
| PLGA-PEG-cRGDfK | Murine U87MG Glioblastoma | - | 5.7 ± 0.7 | 2.7x | 2024 |
| Chitosan (Non-targeted) | Murine HeLa Xenograft | 4.5 ± 0.6 | - | - | 2023 |
| Chitosan-Transferrin | Murine HeLa Xenograft | - | 12.1 ± 1.4 | 2.7x | 2023 |
Table 2: Common Targeting Ligands and Their Receptors
| Ligand | Target Receptor | Common Cancer Types | Conjugation Chemistry |
|---|---|---|---|
| Folic Acid | Folate Receptor (FR-α) | Ovarian, Breast, Lung | NHS-ester to amine, Click chemistry |
| cRGDfK peptide | αvβ3 Integrin | Glioblastoma, Melanoma | Maleimide to thiol, Amide coupling |
| Transferrin | Transferrin Receptor (TfR) | Glioblastoma, Pancreatic | EDC/NHS amidation |
| Anti-HER2 scFv | HER2/neu | Breast, Gastric | Thiol-maleimide, Oxime click |
Key Experimental Protocols
Protocol 1: Synthesis of PLGA-PEG-Folate Nanoparticles
Objective: Prepare actively targeted nanoparticles with a PEG spacer for ligand presentation.
Materials: PLGA-COOH (50:50, 24 kDa), NH2-PEG-COOH (3.4 kDa), Folic Acid, N-Hydroxysuccinimide (NHS), N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC), Dimethyl Sulfoxide (DMSO), Dichloromethane (DCM), Polyvinyl Alcohol (PVA).
Procedure:
- PEG-Folate Conjugate Synthesis: Activate folic acid (10 mg) with EDC (12 mg) and NHS (7 mg) in anhydrous DMSO (2 mL) for 1h. Add to NH2-PEG-COOH (50 mg) in DMSO (1 mL). React overnight under N₂. Purify by dialysis (MWCO 1 kDa) against DMSO, then water. Lyophilize.
- Nanoparticle Formulation: Dissolve PLGA-COOH (50 mg) and PEG-Folate conjugate (5 mg) in DCM (3 mL). Emulsify in 1% w/v PVA solution (10 mL) via probe sonication (70 W, 2 min on ice).
- Solvent Evaporation: Stir emulsion overnight at room temperature to evaporate DCM.
- Purification: Centrifuge nanoparticles at 21,000 × g for 20 min. Wash pellet 3x with DI water. Resuspend in buffer and lyophilize with 5% trehalose as cryoprotectant.
Protocol 2: In Vitro Cellular Uptake Assay (Flow Cytometry)
Objective: Quantify the enhanced cellular internalization of ligand-targeted vs. non-targeted nanoparticles.
Materials: FR-α overexpressing KB cells, Fluorescent dye (e.g., DiI or Coumarin-6)-loaded nanoparticles, Flow cytometry buffer (PBS + 2% FBS), Trypsin-EDTA.
Procedure:
- Seed KB cells in 12-well plates at 2.5 × 10⁵ cells/well. Incubate for 24h.
- Treat cells with DiI-loaded non-targeted (PLGA-PEG) or targeted (PLGA-PEG-Folate) nanoparticles (equivalent dye concentration: 200 ng/mL). Include a group pre-treated with free folic acid (1 mM) for 1h to demonstrate receptor competition.
- Incubate for 2h at 37°C (5% CO₂).
- Wash cells 3x with cold PBS. Detach with trypsin-EDTA, quench with complete media, and centrifuge at 500 × g for 5 min.
- Resuspend cell pellet in 500 µL flow buffer. Analyze using a flow cytometer (Ex/Em: 549/565 nm for DiI). Measure mean fluorescence intensity (MFI) of 10,000 events per sample.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for EPR/Ligand Synergy Research
| Item | Function/Description | Example Vendor/Product |
|---|---|---|
| Functional Polymers | Backbone for nanoparticle formation; provides biocompatibility and drug encapsulation. | PLGA-COOH (Lactel), NH2-PEG-COOH (Creative PEGWorks) |
| Targeting Ligands | Enables specific receptor binding (active targeting). | Folic Acid (Sigma), cRGDfK peptide (GenScript), Human Transferrin (Thermo Fisher) |
| Conjugation Kits | Facilitates covalent attachment of ligands to polymer/particle surface. | EDC/NHS Crosslinking Kit (Thermo Fisher), Maleimide-Thiol Conjugation Kit (Abcam) |
| Fluorescent Probes | For tracking nanoparticle localization and uptake in vitro/in vivo. | DiI, DiO, DIR lipophilic dyes (Thermo Fisher), Coumarin-6 (Sigma) |
| Characterization Inst. | Determines size, charge, and stability of nanoparticles. | Zetasizer Nano (Malvern) for DLS & Zeta Potential |
| Animal Tumor Models | In vivo validation of EPR and targeting efficacy. | Murine 4T1 (breast), U87MG-luc (glioblastoma) xenografts from ATCC/Charles River |
Signaling Pathways in Receptor-Mediated Endocytosis
Diagram Title: Pathway of Ligand-Mediated Endocytosis and Fate
The strategic combination of passive EPR-driven accumulation and ligand-mediated active targeting represents a paradigm shift in oncological nanomedicine, directly addressing the core limitations outlined in the broader thesis on polymeric nanoparticles. This synergistic approach, enabled by precise chemical conjugation and rational design, significantly enhances tumor specificity, cellular uptake, and therapeutic payload delivery. Continued research into tumor microenvironment-specific ligands and stimuli-responsive linkers will further refine this powerful dual-targeting strategy.
Within the paradigm of Enhanced Permeability and Retention (EPR) effect-mediated tumor targeting, the rational design of polymeric nanoparticles (PNPs) hinges on efficient and stable payload incorporation. This technical guide details contemporary methodologies for encapsulating diverse therapeutic agents—small molecules, proteins, and nucleic acids—into PNPs. It provides a comparative analysis of strategies, quantitative encapsulation data, and standardized experimental protocols, serving as a foundational resource for advancing EPR-based drug delivery research.
The EPR effect describes the preferential accumulation of macromolecules and nanoparticles in tumor tissues due to leaky vasculature and impaired lymphatic drainage. Polymeric nanoparticles (e.g., PLGA, PEG-PLGA, chitosan, dendrimers) are prime carriers to exploit this phenomenon. Their efficacy is fundamentally governed by the method of payload incorporation, which dictates loading capacity (LC), encapsulation efficiency (EE), release kinetics, and ultimately, in vivo bioactivity. This guide delineates core strategies tailored to the physicochemical properties of each payload class.
Quantitative Comparison of Incorporation Strategies
Table 1: Comparative Performance of Payload Incorporation Methods
| Payload Type | Primary Method | Typical EE (%) | Typical LC (% w/w) | Key Influencing Factors |
|---|---|---|---|---|
| Hydrophobic Small Molecules | Nanoprecipitation / Single Emulsion | 70 - 95 | 5 - 25 | Polymer-payload affinity, organic solvent choice, aqueous phase surfactant |
| Hydrophilic Small Molecules | Double Emulsion (W/O/W) | 30 - 70 | 1 - 10 | Stability of primary emulsion, diffusion rate, polymer MW |
| Proteins / Peptides | Double Emulsion / Coacervation | 20 - 60 | 1 - 15 | Aqueous phase pH & ionic strength, protein-polymer interaction, process shear stress |
| siRNA / miRNA | Ionic Complexation / Double Emulsion | >90 (complex) 50-80 (encap) | 2 - 10 | N/P ratio, polymer cation density, buffer conditions |
| pDNA / mRNA | Ionic Complexation / Nanocomplexation | >95 (complex) | N/A | Polymer structure, charge balance, steric stabilization (PEGylation) |
Table 2: Common Polymers & Their Payload Affinities
| Polymer | Key Properties | Optimal Payload Match | Notes for EPR |
|---|---|---|---|
| PLGA | Biodegradable, hydrophobic, tunable MW | Hydrophobic drugs, proteins (via W/O/W) | PEGylation enhances circulation time. |
| PEG-PLGA (Diblock) | Amphiphilic, forms micelles/vesicles | Hydrophobic & amphiphilic drugs | PEG shell reduces opsonization. |
| Chitosan | Cationic, mucoadhesive | Nucleic acids, proteins (ionic) | Positive charge may interact with serum. |
| Polyethylenimine (PEI) | High cationic charge density | Nucleic acids (high complexation) | Often modified to reduce cytotoxicity. |
| Dendrimers (PAMAM) | Monodisperse, multivalent surface | Small molecules, nucleic acids, proteins | Size and charge precisely controllable. |
Experimental Protocols for Key Methods
Protocol: Nanoprecipitation for Hydrophobic Small Molecules
Objective: Encapsulate a model hydrophobic drug (e.g., Paclitaxel) into PEG-PLGA nanoparticles. Materials: See Scientist's Toolkit. Procedure:
- Dissolve 50 mg PEG-PLGA and 5 mg paclitaxel in 5 mL of acetone (organic phase).
- Prepare 20 mL of an aqueous phase containing 0.5% (w/v) polyvinyl alcohol (PVA).
- Using a syringe pump set to 1 mL/min, inject the organic phase into the stirred (magnetic stirrer, 600 rpm) aqueous phase.
- Stir the resulting suspension for 3 hours at room temperature to allow for solvent evaporation and nanoparticle hardening.
- Centrifuge at 20,000 x g for 30 min at 4°C. Wash pellet twice with DI water.
- Resuspend nanoparticles in phosphate-buffered saline (PBS) or lyophilize with a cryoprotectant (e.g., 5% trehalose).
- Quantification: Determine EE and LC via HPLC. Dissolve a known amount of nanoparticles in DMSO to release drug, analyze against standard curve. EE% = (Mass of drug in nanoparticles / Initial mass of drug) x 100. LC% = (Mass of drug in nanoparticles / Total mass of nanoparticles) x 100.
Protocol: Double Emulsion (W/O/W) for Proteins
Objective: Encapsulate Bovine Serum Albumin (BSA) as a model protein in PLGA nanoparticles. Procedure:
- Dissolve 100 mg PLGA in 2 mL dichloromethane (DCM) (oil phase).
- Prepare a primary water-in-oil (W/O) emulsion by sonicating (probe sonicator, 30 W, 30 s on ice) 0.5 mL of an aqueous BSA solution (10 mg/mL) with the PLGA/DCM solution.
- This primary W/O emulsion is then immediately poured into 10 mL of an external aqueous phase containing 2% (w/v) PVA and homogenized (high-speed homogenizer, 10,000 rpm, 1 min).
- Stir the resulting W/O/W emulsion overnight to evaporate DCM.
- Centrifuge, wash, and lyophilize as in Protocol 3.1.
- Quantification: Determine EE via Micro-BCA assay. Dissolve nanoparticles in 0.1M NaOH with 1% SDS overnight to degrade polymer and release protein. Measure protein content against a BSA standard.
Protocol: Ionic Complexation for siRNA
Objective: Form stable polyplex nanoparticles with siRNA using a cationic polymer. Procedure:
- Prepare a solution of cationic polymer (e.g., linear PEI, 1 mg/mL in nuclease-free buffer) and siRNA (0.1 mg/mL in the same buffer).
- Calculate the desired N/P ratio (molar ratio of polymer Nitrogen to siRNA Phosphate). For PEI, an N/P of 5-10 is typical.
- Rapidly mix the siRNA solution into the vortexing polymer solution.
- Incubate the mixture at room temperature for 20-30 min to allow polyplex formation.
- Characterization: Analyze particle size and zeta potential via dynamic light scattering (DLS). Confirm complexation via gel retardation assay (run on agarose gel).
Visualization of Strategies & Workflows
Diagram 1: Strategy Selection Workflow for Payload Encapsulation
Diagram 2: Nanoparticle Journey via the EPR Effect
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Payload Incorporation Research
| Reagent / Material | Function / Role | Example Product/Catalog |
|---|---|---|
| PLGA (50:50, acid-terminated) | Biodegradable polymer core for nanoparticle formation. | Lactel Labs B6010-2 |
| mPEG-PLGA Diblock Copolymer | Provides stealth properties for prolonged circulation. | Sigma-Aldrug 774465 |
| Polyvinyl Alcohol (PVA, 87-89% hydrolyzed) | Stabilizing surfactant in emulsion methods. | Sigma-Aldrug 363138 |
| Dichloromethane (DCM) & Acetone (HPLC Grade) | Organic solvents for polymer and drug dissolution. | Fisher Chemical D/1856/17 |
| Linear Polyethylenimine (LPEI, 25 kDa) | Cationic polymer for nucleic acid complexation. | Polysciences 23966 |
| siRNA (e.g., GAPDH targeting) | Model nucleic acid payload for method development. | Dharmacon D-001830-10 |
| Model Proteins (BSA, Lysozyme) | Stable, well-characterized protein payloads. | Sigma-Aldrug A9418 |
| Dialysis Membranes (MWCO 3.5-14 kDa) | Purification of nanoparticles and removal of free payload/solvent. | Spectrum Labs 132720 |
| Cryoprotectant (Trehalose) | Preserves nanoparticle integrity during lyophilization. | Sigma-Aldrug T9531 |
| Dynamic Light Scattering (DLS) Instrument | Measures nanoparticle hydrodynamic size and zeta potential. | Malvern Zetasizer Nano ZS |
Within the broader thesis on the Enhanced Permeability and Retention (EPR) effect and polymeric nanoparticle research, stimuli-responsive polymers represent a pivotal advancement. The EPR effect facilitates the passive accumulation of nanocarriers in tumor tissues, but it lacks specificity for cellular internalization and intracellular drug release. Stimuli-responsive "smart" polymers address this by enabling controlled, site-specific payload release in response to pathological microenvironmental cues, thereby enhancing therapeutic efficacy and reducing systemic toxicity. This technical guide details three principal endogenous triggers: pH, redox potential, and enzyme activity.
Core Mechanisms and Polymer Chemistries
pH-Sensitive Systems
Tumor microenvironments (TME) and endo/lysosomal compartments exhibit acidic pH (~6.5-7.2 in TME, ~4.5-6.0 in endosomes/lysosomes) compared to physiological pH (7.4). Polymers exploit this gradient via:
- Ionizable Groups: Polymers containing tertiary amines (e.g., poly(β-amino esters) - PBAEs, poly(histidine)) protonate in acidic environments, leading to swelling or dissolution.
- Acid-Labile Linkers: Covalent bonds such as hydrazone, acetal, ketal, and cis-aconityl, which hydrolyze at low pH, cleaving drug-polymer conjugates.
Redox-Sensitive Systems
The intracellular milieu has a high glutathione (GSH) concentration (2-10 mM), 100-1000 times higher than the extracellular space (~2-20 μM). This differential enables cleavage of disulfide bonds (-S-S-) incorporated into polymer backbones, side chains, or as cross-linkers.
Enzyme-Sensitive Systems
Overexpressed enzymes in disease sites (e.g., matrix metalloproteinases - MMPs, phospholipases, glycosidases in tumors) catalyze the cleavage of specific peptide or other sequences grafted onto polymers.
Table 1: Characteristic Triggers in Physiological vs. Pathological Microenvironments
| Stimulus | Physiological Condition | Pathological/Target Condition (e.g., Tumor) | Key Responsive Chemical Motif |
|---|---|---|---|
| pH | 7.4 (blood, extracellular) | 6.5-7.2 (TME), 4.5-6.0 (endo/lysosome) | Tertiary amines, Hydrazone, Acetal |
| Redox Potential | Low [GSH] (~2-20 μM extracellular) | High [GSH] (2-10 mM intracellular) | Disulfide bond (-S-S-) |
| Enzymes | Low/regulated protease activity | Overexpression (e.g., MMP-2/9, Cathepsin B) | Peptide sequences (e.g., GFLG, PLGLAG) |
Table 2: Representative Stimuli-Responsive Polymers and Drug Release Kinetics
| Polymer System | Stimulus | Trigger Condition | Drug Payload | Release Half-time/Profile (Approx.) | Reference Key |
|---|---|---|---|---|---|
| PBAE-hydrazone-DOX | pH | pH 5.0 vs. 7.4 | Doxorubicin (DOX) | ~12 h (pH 5.0) vs. <10% @ 24h (pH 7.4) | [1] |
| PEG-SS-PCL micelle | Redox | 10 mM GSH vs. 10 μM GSH | Paclitaxel (PTX) | >80% @ 12h (10mM GSH) vs. <20% @ 12h (10μM) | [2] |
| PEG-Peptide-PCL | Enzyme | 1 μg/mL MMP-2 | DOX | >70% @ 24h (w/ MMP-2) vs. <15% @ 24h (w/o) | [3] |
Experimental Protocols for Key Evaluations
Protocol: In Vitro Drug Release under Differential pH
Objective: To quantify the pH-responsive release profile of a drug-loaded polymeric nanoparticle. Materials: Dialysis bag (MWCO appropriate for polymer), release media (PBS at pH 7.4, 6.5, and 5.0), water bath shaker. Procedure:
- Dispense 1 mL of nanoparticle suspension (e.g., 2 mg/mL drug-loaded PBAE NPs) into a pre-hydrated dialysis bag.
- Seal the bag and immerse it in 200 mL of pre-warmed release medium (37°C) under gentle agitation (100 rpm).
- At predetermined time points (0.5, 1, 2, 4, 8, 12, 24, 48 h), withdraw 1 mL of external medium and replace with an equal volume of fresh pre-warmed medium.
- Quantify the drug concentration in the withdrawn samples using HPLC or UV-Vis spectrophotometry (e.g., DOX: λex 480 nm, λem 590 nm).
- Calculate cumulative release percentage and plot vs. time.
Protocol: Confirming Redox-Responsive Disassembly via DLS
Objective: To monitor the size change of disulfide-crosslinked nanoparticles upon GSH treatment. Materials: Dynamic Light Scattering (DLS) instrument, GSH stock solution (prepared in degassed PBS), nanoparticle suspension. Procedure:
- Measure the initial hydrodynamic diameter (D_h) and polydispersity index (PDI) of the nanoparticle suspension (e.g., 0.5 mg/mL in PBS).
- Add GSH to the nanoparticle suspension to achieve final concentrations simulating extracellular (e.g., 10 μM) and intracellular (e.g., 10 mM) conditions.
- Incubate at 37°C. Measure D_h and PDI at set intervals (e.g., 0, 15, 30, 60, 120 min).
- A significant increase in D_h or a decrease in scattering intensity indicates disassembly due to disulfide cleavage.
Protocol: Validating Enzyme-Specific Cleavage
Objective: To demonstrate enzyme-triggered payload release or morphological change. Materials: Target enzyme (e.g., MMP-2), specific substrate peptide-linked fluorophore (e.g., DABCYL/Gly-Pro-Leu-Gly-Val-Arg-Gly/EDANS), fluorometer. Procedure:
- Incubate enzyme-sensitive nanoparticles (or a model peptide-polymer conjugate) with the target enzyme in its optimal activity buffer.
- For fluorometric assays, monitor the increase in fluorescence signal (e.g., EDANS emission at 490 nm upon excitation at 340 nm) due to substrate cleavage over time.
- Include control groups without enzyme and with enzyme + inhibitor (e.g., EDTA for MMPs).
- Confirm cleavage products via HPLC or mass spectrometry.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions & Materials
| Item | Function/Brief Explanation |
|---|---|
| Poly(β-amino ester) (PBAE) | A biodegradable cationic polymer with tertiary amines providing pH-dependent solubility/swelling. |
| D,L-Dithiothreitol (DTT) | A strong reducing agent used as a substitute for GSH in proof-of-concept redox-responsiveness experiments. |
| Glutathione (GSH), Reduced | The primary endogenous reducing agent used to simulate intracellular reductive conditions. |
| Matrix Metalloproteinase-2 (MMP-2) | A key overexpressed enzyme in tumor metastasis used to trigger enzyme-sensitive systems. |
| Cy5.5 or DIR Near-Infrared Dye | Hydrophobic dyes for encapsulating into nanoparticles for in vivo fluorescence imaging of biodistribution and EPR-mediated tumor accumulation. |
| Dialysis Tubing (various MWCO) | For purifying nanoparticles and conducting in vitro drug release studies via the dialysis method. |
| Citrate-Phosphate Buffers (pH 4.0-7.4) | Provide precise pH conditions for testing pH-responsive behavior in vitro. |
| CCK-8 Cell Viability Assay Kit | Standard reagent for assessing in vitro cytotoxicity of drug-loaded nanoparticles against cancer cell lines. |
Visualizations
Title: pH-Sensitive Nanoparticle Drug Release Pathway
Title: Redox-Triggered Disassembly and Release
Title: Workflow for Enzyme-Responsive System Development
References (Key Examples): [1] Lynn, D.M., et al. (2001). J. Am. Chem. Soc. [2] Sun, H., et al. (2014). ACS Nano. [3] Zhu, L., et al. (2012). Proc. Natl. Acad. Sci. U.S.A.
The translation of polymeric nanoparticles (PNPs) designed to exploit the Enhanced Permeability and Retention (EPR) effect from laboratory discovery to clinical application is a pivotal challenge. This guide outlines the critical technical and regulatory considerations for achieving reproducible Good Manufacturing Practice (GMP) manufacturing of PNPs, ensuring that promising preclinical results translate into safe and effective clinical therapies.
Core Challenges in Scaling EPR-Based PNPs
The scale-up of PNPs must preserve the critical quality attributes (CQAs) that enable the EPR effect: particle size (typically 10-200 nm), narrow size distribution (PDI < 0.2), surface charge (near-neutral or slightly negative for prolonged circulation), drug loading efficiency, and sterility.
Table 1: Key CQAs for EPR-Effect PNPs and Scaling Challenges
| Critical Quality Attribute (CQA) | Target Range (Preclinical) | Primary Scaling Challenge | Impact on EPR/ Efficacy |
|---|---|---|---|
| Mean Hydrodynamic Diameter | 20 - 150 nm | Aggregation, shear stress during mixing | Determines vascular extravasation and tumor penetration |
| Polydispersity Index (PDI) | < 0.2 | Inconsistent mixing leading to broad nucleation | Affects pharmacokinetic uniformity and biodistribution |
| Zeta Potential | -10 to +10 mV (stealth) | Changes in solvent/ionic environment | Influences protein corona formation and circulation half-life |
| Drug Loading (%) | > 5% (often > 10%) | Altered kinetics of polymer-drug assembly | Directly impacts therapeutic dose and potential efficacy |
| Encapsulation Efficiency (%) | > 80% | Drug partitioning changes at larger volumes | Impacts cost, potency, and burst release profile |
| Sterility & Endotoxins | Sterile, EU/mL < 0.25 | Aseptic processing vs. terminal sterilization | Critical for patient safety; some PNPs cannot be autoclaved |
From Bench-Side Synthesis to GMP Processes
Bench-Side Protocol: Nanoprecipitation of PLGA-PEG PNPs
- Materials: PLGA-PEG copolymer (e.g., RESOMER), active pharmaceutical ingredient (API), acetone (organic phase), deionized water (aqueous phase), magnetic stirrer, syringe pump, sonicator (bath or probe).
- Method:
- Dissolve PLGA-PEG and API in acetone (organic phase) at a defined concentration (e.g., 10 mg polymer/mL).
- Filter the organic solution through a 0.22 µm PTFE syringe filter.
- Place the aqueous phase (water) under high-speed magnetic stirring (e.g., 800 rpm).
- Using a syringe pump, add the organic phase dropwise (e.g., 1 mL/min) into the aqueous phase.
- Stir for 3-4 hours at room temperature to evaporate acetone.
- Concentrate and diafilter using tangential flow filtration (TFF) with a 100 kDa membrane against phosphate-buffered saline (PBS).
- Characterize CQAs (size, PDI, zeta potential by DLS; drug loading by HPLC).
Scale-Up & GMP Considerations:
- Process Parameter Translation: Stirring speed must be translated to scalable impeller systems maintaining equivalent shear stress and mixing efficiency (Reynolds number). Dropwise addition is replaced with controlled, high-pressure mixing or static mixers.
- Material Changes: Reagent-grade solvents must be replaced with GMP-grade materials. Polymers must be sourced from GMP-compliant vendors with certificates of analysis (CoA).
- Closed Systems: Open beaker setups are replaced with closed, stainless-steel or single-use bioreactor/ mixing systems to ensure aseptic processing.
- Purification: Lab-scale dialysis is replaced by robust, scalable TFF, enabling consistent buffer exchange, concentration, and endotoxin control.
- Process Analytical Technology (PAT): Implement in-line monitoring (e.g., focused beam reflectance measurement for particle size, UV for drug concentration) for real-time quality control versus offline sampling.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for PNP Development & Scale-Up
| Item | Function & Importance | GMP Considerations |
|---|---|---|
| GMP-Grade Polymers (e.g., PLGA, PLGA-PEG, mPEG-PLGA) | The backbone of nanoparticle formation; determines biodegradability, drug release, and stealth properties. | Require Drug Master File (DMF) or equivalent regulatory support. Vendor must provide full traceability and CoA for identity, purity, Mw, residual solvents. |
| High-Purity API | The therapeutic agent to be encapsulated. | Must meet pre-defined specifications for identity, potency, and impurities. Compatibility with polymer and process is critical. |
| GMP-Grade Solvents (e.g., Acetone, Ethyl Acetate, DMSO) | Used to dissolve polymer and API in the organic phase. | Must be sterile-filtered or come in sterile single-use containers. Residual solvent levels in final product must meet ICH Q3C guidelines. |
| TFF/UF Systems & Membranes | For concentration, buffer exchange, and removal of residual solvents/unencapsulated API. | System must be validated for consistency and sterility. Membrane molecular weight cutoff (MWCO) and material compatibility must be defined. |
| Sterile Single-Use Assemblies (bags, tubing, connectors, filters) | For fluid transfer, mixing, and filtration in a closed system. | Reduce cross-contamination risk and eliminate cleaning validation. Must pass USP <87>/<88> biocompatibility tests. |
| In-Process Control Standards (e.g., size standards, endotoxin standards) | For calibrating and validating analytical equipment (DLS, HPLC, LAL assay). | Traceable to national/international standards. Essential for assay qualification/validation. |
Critical Regulatory & Analytical Pathway
A Quality by Design (QbD) framework is essential. This involves defining a Target Product Profile (TPP), identifying CQAs, and establishing the link between critical material attributes (CMAs), critical process parameters (CPPs), and the CQAs.
Diagram Title: QbD Framework for PNP Process Development
Key Experimental Workflow for Process Development
The following workflow is critical for establishing a scalable, reproducible process.
Diagram Title: PNP Process Development & Scale-Up Workflow
Stability and Sterilization Considerations
Long-term stability (real-time and accelerated) is required for clinical trials. Terminal sterilization (autoclaving) is often incompatible with PNPs, making aseptic processing via filtration (0.22 µm) mandatory. All processes and excipients must control endotoxin levels from the start.
Table 3: Common Sterilization Methods for PNPs
| Method | Typical Application | Key Risk for PNPs | Mitigation Strategy |
|---|---|---|---|
| Aseptic Filtration | Primary method for heat-labile PNPs. | Polymer/particle adsorption to filter; shear-induced aggregation; inability to filter >200 nm particles. | Filter compatibility studies; use of low-protein-binding filters (e.g., PES). |
| Terminal Autoclaving | Standard for many parenterals. | Particle aggregation, drug degradation, polymer hydrolysis acceleration. | Generally avoided. If necessary, extensive pre- and post-sterilization CQA testing. |
| Gamma Irradiation | For terminal sterilization of finished product in vial. | Polymer chain scission or cross-linking, radical-induced API degradation. | Requires detailed dosage studies and protective excipient screening. |
Successful GMP manufacturing of EPR-targeted PNPs hinges on early and deliberate planning for scale-up. By adopting a QbD approach, meticulously translating process parameters, and implementing robust analytical controls, researchers can bridge the gap between promising bench-side data and reproducible, clinically viable nanomedicines. The integration of scalable unit operations and PAT from the earliest development stages is non-negotiable for ensuring that the therapeutic potential demonstrated in preclinical models is faithfully delivered to patients.
Navigating Clinical Translation Hurdles: Overcoming EPR Limitations and Nanoparticle Challenges
The Enhanced Permeability and Retention (EPR) effect remains a cornerstone for the passive tumor targeting of nanomedicines, particularly polymeric nanoparticles. However, significant clinical translation challenges persist, largely due to profound inter- and intra-tumor heterogeneity. This technical guide, framed within ongoing thesis research on EPR optimization, analyzes the physiological barriers contributing to variable EPR and details contemporary strategies to overcome them. We focus on advanced polymeric nanoparticle designs and adjunctive therapies that aim to homogenize and enhance drug delivery across heterogeneous tumor microenvironments.
Tumor heterogeneity manifests at multiple levels: vascular density, perfusion, permeability, interstitial fluid pressure (IFP), stromal composition, and cellular phenotype. This variability leads to the non-uniform "EPR effect," where nanoparticle accumulation and penetration are inefficient and unpredictable, compromising therapeutic efficacy. This document consolidates current research on engineering solutions to normalize the tumor microenvironment and engineer smarter nanoparticles, thereby converting the heterogeneous EPR into a more reliable delivery phenomenon.
Physiological Determinants of Heterogeneous EPR
Quantitative Analysis of Tumor Microenvironment Variability
The following table summarizes key parameters that contribute to EPR heterogeneity, based on recent in vivo studies.
Table 1: Quantitative Parameters of Tumor Heterogeneity Affecting EPR
| Parameter | Typical Range in Solid Tumors | Impact on EPR | Measurement Technique |
|---|---|---|---|
| Vascular Permeability (Ktrans) | 0.01 - 0.5 min⁻¹ | Directly determines initial extravasation. | Dynamic Contrast-Enhanced MRI (DCE-MRI) |
| Interstitial Fluid Pressure (IFP) | 10 - 100 mmHg (vs. ~0 in normal tissue) | Creates outward convective flow, hindering inward diffusion. | Wick-in-needle, Micropressure systems |
| Collagen Density | 1.5x - 8x higher than normal tissue | Increases matrix stiffness, blocks nanoparticle diffusion. | Second Harmonic Generation (SHG) microscopy |
| Hyaluronic Acid Content | Up to 10 µg/mg tissue | Contributes to high IFP & steric hindrance. | Mass spectrometry, ELISA of biopsies |
| Tumor Blood Flow | Highly heterogeneous, often <10% of cardiac output | Limits nanoparticle delivery to tumor core. | Laser Doppler flowmetry, DCE-MRI |
| Macrophage Density (TAMs) | 5-40% of tumor mass | Can sequester and clear nanoparticles. | Immunohistochemistry (IHC) for CD68/CD206 |
Signaling Pathways Governing EPR Determinants
The dysregulated vascular and stromal environment is controlled by complex signaling networks.
Diagram Title: Signaling Pathways Driving EPR Heterogeneity
Strategic Approaches to Improve EPR Uniformity
Tumor Microenvironment Priming (Normalization)
This strategy uses pharmacological agents transiently to "normalize" pathological tumor features, creating a window of improved EPR.
Experimental Protocol: Evaluating Anti-Angiogenic Priming
Objective: To assess how pre-treatment with a vascular normalization agent (e.g., anti-VEGFR2 antibody DC101) improves the distribution and efficacy of subsequently administered polymeric nanoparticles.
Materials:
- Murine tumor model (e.g., 4T1 breast carcinoma, CT26 colon carcinoma).
- Anti-VEGFR2 antibody (DC101) or small-molecule TKI (e.g., Sunitinib).
- Fluorescently labeled polymeric nanoparticles (e.g., PLGA-PEG nanoparticles loaded with DiR dye).
- In vivo imaging system (IVIS) or similar.
- Immunohistochemistry reagents for CD31 (vessels), α-SMA (pericytes), Hypoxyprobe.
Procedure:
- Tumor Implantation & Growth: Inoculate tumor cells subcutaneously into mice. Allow tumors to grow to ~100 mm³.
- Priming Treatment: Randomize mice into two groups: (i) Control (PBS) and (ii) Normalization (DC101, 40 mg/kg, i.p.). Administer priming dose every 3 days.
- Monitoring Window: On days 2, 4, 7, and 10 after first priming dose: a. Sacrifice a cohort from each group. b. Harvest tumors, freeze-section. c. Perform IHC for CD31 and α-SMA. Calculate vessel pericyte coverage index (α-SMA⁺ area / CD31⁺ area). d. Inject Hypoxyprobe 1 hr before sacrifice to quantify hypoxic fraction.
- Nanoparticle Administration & Imaging: On the identified "optimal normalization day" (e.g., day 4-5, based on step 3), inject fluorescent nanoparticles (5 mg/kg, i.v.) into a new cohort of primed and control mice.
- Quantification: At 24h post-injection, image mice ex vivo using IVIS to quantify total tumor fluorescence. Process tumors for frozen sections and use confocal microscopy to measure nanoparticle penetration depth (distance from vessel wall) and distribution homogeneity.
Expected Outcome: The normalization group should show increased pericyte coverage, reduced hypoxia, more uniform and deeper nanoparticle distribution, and higher overall tumor accumulation compared to the control.
Stroma-Modulating Strategies
High stromal density is a major barrier to penetration. Strategies include enzymatic degradation or inhibition of stromal component synthesis.
Table 2: Stroma-Modulating Agents for EPR Enhancement
| Agent Class | Example(s) | Target | Effect on EPR Determinants | Key Consideration |
|---|---|---|---|---|
| Enzymatic Degrader | PEGylated Hyaluronidase (PEGPH20) | Hyaluronic Acid | Reduces IFP, decompresses vessels, increases diffusion. | Can increase metastasis risk; requires careful dosing window. |
| Collagen Synthesis Inhibitor | Tranilast, Losartan | TGF-β, Angiotensin II | Reduces collagen I/III deposition, decreases matrix stiffness. | Long-term treatment needed; effects are gradual. |
| FAK Inhibitor | Defactinib | Focal Adhesion Kinase | Reduces stromal fibrosis and cancer cell survival. | Impacts cell adhesion signaling broadly. |
Advanced Polymeric Nanoparticle Engineering
Modern designs move beyond passive accumulation to actively overcome barriers.
Size- and Shape-Shifting Nanoparticles
Protocol: Fabrication of pH-Sensitive Size-Shifting Nanoparticles
Objective: To synthesize polymeric nanoparticles that are large (~100 nm) for prolonged circulation and high initial tumor accumulation, but shrink to smaller particles (~20 nm) in the acidic tumor microenvironment to enhance penetration.
Materials:
- Polymer 1: PLGA-b-PEG (acid-insensitive, MW 50k-10k).
- Polymer 2: PHis-b-PEG (Polyhistidine-b-PEG, pH-sensitive, MW 20k-5k).
- Dichloromethane (DCM), Polyvinyl Alcohol (PVA), Dialysis tubing.
- Fluorescent dye (e.g., Cy5.5 NHS ester) for labeling.
Procedure:
- Polymer Synthesis & Conjugation: Synthesize or procure PLGA-PEG and PHis-PEG. Conjugate Cy5.5 to the PEG terminus of a portion of each polymer via NHS chemistry.
- Nanoparticle Formulation: Use a double emulsion solvent evaporation method. a. Dissolve PLGA-PEG and PHis-PEG at a 70:30 w/w ratio in DCM. b. Add the organic phase to an aqueous 1% PVA solution and emulsify using a probe sonicator (30% amplitude, 1 min) on ice. c. Pour the primary emulsion into a larger volume of 0.3% PVA and stir overnight to evaporate DCM. d. Collect nanoparticles by centrifugation (20,000 g, 30 min), wash 3x with water, and lyophilize.
- Characterization: Measure size and zeta potential in buffers at pH 7.4 and pH 6.5 using Dynamic Light Scattering (DLS). Confirm size shift after 2h incubation at target pH.
- In Vitro Penetration Assay: a. Create a 3D tumor spheroid from cancer cells (e.g., MCF-7). b. Incubate spheroids with size-shifting nanoparticles or control static-sized nanoparticles. c. After 24h, image spheroids using confocal microscopy. Quantify fluorescence intensity from the rim to the core (radial profile) to assess penetration.
Multi-Stage and Transformable Delivery Systems
These systems sequentially overcome different barriers.
Diagram Title: Workflow of a Multi-Stage Polymeric Delivery System
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for EPR Heterogeneity Research
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| PEGylated Polymeric Nanoparticles | Gold-standard, long-circulating control particle for baseline EPR studies. | 100 nm Fluorescent PS-PEG Nanoparticles (e.g., Nanocs, PG-FF100). |
| MMP-Sensitive Peptide Linker | To create stimuli-responsive, transformable nanoparticles. | GPLGIAGQ peptide (custom synthesis from PeptideGen). |
| Recombinant VEGF / Anti-VEGF | To manipulate vascular permeability and maturity in in vitro models. | Mouse VEGF-165 Recombinant Protein (R&D Systems, 493-MV). |
| Hyaluronidase (PEGPH20-like) | For in vitro and in vivo stroma modulation experiments. | Recombinant Human PH20 Hyaluronidase (Sigma, SRP6310). |
| Hypoxia Probe | To quantify tumor hypoxia, a key driver of heterogeneity. | Hypoxyprobe-1 (Pimonidazole HCl) Kit (Hypoxyprobe Inc). |
| Matrigel (High Protein) | To create a dense, in-vivo-like extracellular matrix for 3D penetration assays. | Corning Matrigel Matrix, Phenol Red-free (Corning, 356237). |
| In Vivo Imaging Dye | For non-invasive tracking of nanoparticle biodistribution. | DIR near-infrared lipophilic dye (Thermo Fisher, D12731). |
| Pressure Catheter System | For direct measurement of Interstitial Fluid Pressure (IFP). | Millar Mikro-Tip SPR-1000 catheter (ADInstruments). |
Addressing tumor heterogeneity is not a single-strategy endeavor. The future lies in precision nanomedicine: using imaging biomarkers (e.g., from DCE-MRI, PET) to stratify patients based on their "EPR phenotype," followed by tailored combination of microenvironment normalization and advanced, intelligent polymeric nanoparticles. Continued research must focus on understanding the temporal dynamics of these strategies and developing robust, scalable manufacturing processes for complex, multi-functional nanocarriers. By systematically deconvoluting and targeting the sources of heterogeneity, the promise of the EPR effect can be more reliably harnessed for effective cancer therapy.
The Enhanced Permeability and Retention (EPR) effect has long been the cornerstone for designing polymeric nanoparticles (PNPs) for tumor targeting. The premise is that nanoparticles of a specific size (typically 10-200 nm) can passively accumulate in tumor tissue due to leaky vasculature and impaired lymphatic drainage. However, upon intravenous administration, PNPs are immediately immersed in a complex biological milieu, where proteins and other biomolecules adsorb onto their surface, forming a dynamic layer known as the "protein corona." This corona fundamentally masks the engineered surface, altering the nanoparticle's intrinsic physicochemical properties and biological identity. This whitepaper examines the conundrum of the protein corona, detailing its impact on PNP behavior within the thesis of optimizing the EPR effect for targeted drug delivery.
Composition and Dynamics of the Protein Corona
The protein corona is not a static monolayer but a dynamic, evolving structure comprising a "hard corona" of tightly bound, high-affinity proteins and a "soft corona" of rapidly exchanging, lower-affinity proteins. Its composition depends on nanoparticle core material, surface chemistry (PEGylation, charge, functional groups), size, shape, and the biological fluid (e.g., plasma vs. serum).
Table 1: Key Proteins Commonly Identified in the Hard Corona of Polymeric Nanoparticles and Their Implications
| Protein | Approx. Molecular Weight (kDa) | Typical Abundance in Corona | Potential Impact on Nanoparticle Fate |
|---|---|---|---|
| Albumin | 66.5 | High (30-60%) | Can promote "stealth" properties but may also facilitate transport to certain tissues. |
| Apolipoproteins (ApoA-I, ApoE) | 28-34 | Variable (5-30%) | Critical for cellular recognition; ApoE can mediate brain targeting or hepatic clearance. |
| Immunoglobulins (IgG) | 150 | Moderate (5-15%) | Can trigger opsonization and recognition by the mononuclear phagocyte system (MPS). |
| Fibrinogen | 340 | Low-Moderate | Strong opsonin; promotes macrophage uptake and can affect coagulation. |
| Complement Proteins (C3, C1q) | ~185-410 | Low (1-10%) | Activates complement system, leading to inflammatory responses and clearance. |
Impact on Physicochemical Properties
The adsorbed corona layer induces significant changes to the nanoparticle's measurable properties.
Table 2: Alteration of Nanoparticle Properties Post-Corona Formation
| Property | Pre-Corona Measurement | Post-Corona Measurement | Method of Analysis |
|---|---|---|---|
| Hydrodynamic Diameter | e.g., 100 nm | Increase of 10-30 nm | Dynamic Light Scattering (DLS) |
| Surface Charge (Zeta Potential) | e.g., -30 mV | Shifts towards plasma protein charge (-10 to -15 mV) | Phase Analysis Light Scattering |
| Surface Plasmon Resonance (for some) | Specific peak | Red-shift or damping | UV-Vis Spectroscopy |
| Surface Functional Group Availability | High | Significantly reduced | Fluorescence quenching assays |
The Targeting Paradox: Masking and Unmasking
The protein corona often obscures active targeting ligands (e.g., antibodies, peptides) attached to the PNP surface, diminishing their specific binding to target receptors (e.g., EGFR, HER2). This creates a paradox where targeting specificity, designed to augment the EPR effect, is lost in vivo. However, recent research suggests the corona can also create a new biological identity that may lead to de facto targeting, for instance, through apolipoprotein-mediated pathways.
Experimental Protocol: Assessing Targeting Ligand Availability Post-Corona
- Objective: To quantify the shielding of surface-conjugated RGD peptides by the human plasma protein corona.
- Materials: RGD-conjugated PLGA nanoparticles, fresh human platelet-poor plasma, fluorescently labeled anti-RGD monoclonal antibody.
- Method:
- Incubate PNPs (1 mg/mL) with 50% human plasma in PBS at 37°C for 1 hour.
- Ultracentrifuge at 100,000 x g for 45 min to pellet corona-coated PNPs. Wash gently twice with PBS.
- Resuspend corona-coated PNPs and bare PNPs (control) in buffer.
- Incubate with fluorescent anti-RGD antibody (1:100 dilution) for 1 hour at 4°C.
- Wash and analyze fluorescence intensity per particle via flow cytometry (nanoparticle tracking flow cytometer) or measure solution fluorescence.
- Analysis: A reduction in fluorescence signal in the corona sample indicates ligand masking. Calculate the percentage of accessible ligands relative to the bare PNP control.
Diagram Title: Experimental Workflow for Assessing Ligand Masking by Protein Corona
Key Experimental Protocols in Corona Research
Protocol 1: Isolation and Characterization of the Hard Protein Corona
- Incubation: Incubate purified PNPs (0.5-1 mg/mL) with undiluted or 50% human plasma/serum in physiological buffer (e.g., PBS) at 37°C for 1 hour with gentle agitation.
- Separation: Separate nanoparticle-protein complexes from unbound proteins via:
- Ultracentrifugation: >100,000 x g, 45-60 min, 4°C. Wash pellet gently.
- Size Exclusion Chromatography (SEC): Using columns like Sepharose CL-4B.
- Magnetic Separation: For magnetic core PNPs.
- Protein Elution & Analysis: Elute hard corona proteins using 1-4% SDS in Laemmli buffer at 95°C for 10 min. Analyze via:
- SDS-PAGE: For visual profiling.
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): For definitive protein identification and semi-quantification (label-free or isotopic).
Protocol 2: In Vitro Cellular Uptake Study in the Presence of Corona
- Corona Formation: Form corona on fluorescently labeled PNPs as in Protocol 1.
- Cell Culture: Use relevant cell lines (e.g., HeLa, RAW 264.7 macrophages). Seed in 24-well plates.
- Dosing: Apply bare and corona-coated PNPs at equivalent particle number concentrations (e.g., 50 µg/mL) to cells in serum-free medium.
- Incubation: Incubate for 2-6 hours at 37°C.
- Analysis: Wash, trypsinize, and analyze cell-associated fluorescence via flow cytometry. Confirm with confocal microscopy.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Reagent | Function & Application in Corona Studies |
|---|---|
| Poly(lactic-co-glycolic acid) (PLGA) Nanoparticles | The benchmark biodegradable, FDA-approved polymer for forming PNPs; available commercially in various sizes, charges, and with surface carboxyl or amine groups. |
| Methoxy-PEG-amine (mPEG-NH₂) / PEGylation Kits | For creating stealth coatings to study how PEG density and chain length influence corona composition and cellular uptake. |
| Human Platelet-Poor Plasma (PPP) or Serum | The most physiologically relevant protein source for in vitro corona formation studies. Prefer PPP to include fibrinogen. |
| Pre-formed Fluorescent Nanospheres (e.g., PS, SiO₂) | Model nanoparticles with uniform size and strong fluorescence, used as standards to dissect material-specific corona effects. |
| Protease Inhibitor Cocktails (Tablets/Liquid) | Added to plasma during incubation to prevent protein degradation and ensure corona composition integrity. |
| BCA or Micro-BCA Protein Assay Kit | For colorimetric quantification of total protein adsorbed onto the nanoparticle surface after corona isolation. |
| LC-MS/MS Grade Trypsin | For digesting corona proteins into peptides for mass spectrometric identification and quantification. |
| Dynabeads or Similar Magnetic Beads | Magnetic core particles that enable rapid, low-shear separation of corona-coated nanoparticles from free plasma. |
Diagram Title: The Corona Conundrum: Diverting Nanoparticles from Intended Fate
The protein corona presents a formidable conundrum that bridges the nanomaterial design phase and the complex physiological reality. For research centered on the EPR effect and polymeric nanoparticles, ignoring the corona leads to a significant translational gap. The future lies in moving from viewing the corona as a problem to be prevented, towards understanding and exploiting it. Strategies include pre-forming a "custom" corona in vitro, designing surface chemistries that recruit beneficial proteins (e.g., ApoE for brain delivery), or developing dynamic surfaces that shed the initial corona to reveal targeting ligands at the disease site. Mastery of the corona is essential for transforming the promise of nanoparticle-mediated targeted drug delivery into a clinical reality.
The Enhanced Permeability and Retention (EPR) effect remains a cornerstone concept in nanoparticle-based drug delivery. Within this framework, optimizing pharmacokinetics (PK) and biodistribution is paramount for achieving therapeutic efficacy. This guide details the technical strategies and experimental approaches for balancing the critical triad of circulation longevity, target site accumulation, and eventual clearance, with a focus on polymeric nanoparticles.
Core Principles: The Balancing Act
The fate of an intravenously administered polymeric nanoparticle is governed by a series of competing physiological processes. Long circulation time (increased by stealth properties) enhances the probability of extravasation at pathological sites with leaky vasculature (EPR effect). However, this must be balanced against the need for eventual clearance to prevent long-term toxicity. The design parameters directly influence this balance.
Table 1: Key Nanoparticle Design Parameters and Their Impact on PK/BD
| Parameter | Primary Effect on Circulation | Primary Effect on Accumulation (EPR) | Primary Effect on Clearance |
|---|---|---|---|
| Size | <6 nm: Rapid renal clearance. 10-150 nm: Optimal for long circulation. >200 nm: Increased MPS uptake. | 20-200 nm: Optimal for tumor accumulation via EPR. | >6 nm: Avoids renal clearance. Liver/spleen clearance dominates. |
| Surface Charge (Zeta Potential) | Neutral/slightly negative (-10 to +10 mV): Reduces protein opsonization, long circulation. | Neutral/slightly negative: Promotes diffusion through interstitial matrix. | Highly positive/negative: Increases MPS uptake, rapid clearance. |
| Hydrophilicity & Stealth (e.g., PEGylation) | High: Dramatically reduces opsonization, extends half-life (from minutes to hours/days). | High: Can hinder cellular uptake at target site ("PEG dilemma"). | High: Can reduce hepatic clearance, potentially increasing long-term retention. |
| Surface Ligands (Targeting) | Can increase immunogenicity and clearance if not shielded. | Increases specific cellular internalization at target site (active targeting). | Can alter clearance pathways depending on receptor expression in clearance organs. |
Experimental Protocols for Characterization
Protocol: Measuring Plasma Circulation Half-Life
Objective: Determine the blood clearance kinetics of radiolabeled or fluorescently labeled polymeric nanoparticles.
- Nanoparticle Labeling: Synthesize nanoparticles incorporating a traceable label (e.g., ^3H, ^125I, Cy5.5, DiR). Ensure labeling does not alter surface properties.
- Administration: Intravenously inject a known dose (e.g., 5 mg/kg nanoparticle in 100 µL saline) into rodent tail vein (n=5-8 per group).
- Blood Sampling: Collect small blood samples (e.g., 20 µL) from the retro-orbital plexus or tail nick at predefined time points (e.g., 2 min, 15 min, 30 min, 1, 2, 4, 8, 12, 24, 48 h).
- Quantification: For radioactive labels, measure counts in a gamma counter. For fluorescent labels, lyse blood samples, extract dye, and measure fluorescence intensity against a standard curve.
- Pharmacokinetic Analysis: Fit plasma concentration-time data using a two-compartment model (e.g., using PK solver software). Calculate key parameters: AUC (Area Under Curve), t1/2α (distribution half-life), t1/2β (elimination half-life), and Clearance (CL).
Protocol: Quantitative Biodistribution Study
Objective: Quantify nanoparticle accumulation in target (e.g., tumor) and major clearance organs.
- Preparation: Use labeled nanoparticles as in 3.1.
- Dosing & Sacrifice: Administer IV dose. Euthanize animals at selected endpoints (e.g., 4 h, 24 h, 96 h). Perfuse with saline via the left ventricle to clear blood from organs.
- Organ Harvest: Excise organs of interest: tumor, liver, spleen, kidneys, lungs, heart, and a blood sample. Weigh each organ.
- Analysis:
- Radioactive: Homogenize organs, digest aliquots, and count radioactivity. Express data as % Injected Dose per Gram of tissue (%ID/g) and % Injected Dose per whole organ (%ID/organ).
- Fluorescent: For near-infrared dyes, image whole organs using an ex vivo imaging system (IVIS). Quantify fluorescence intensity and convert to %ID/g using a calibration curve from spiked control organs.
- Data Interpretation: High tumor-to-liver/spleen ratios indicate successful targeting and avoidance of the Mononuclear Phagocyte System (MPS).
Table 2: Typical Biodistribution Profile of Optimized Polymeric Nanoparticles (e.g., PEG-PLGA) in Tumor-Bearing Mice at 24h Post-Injection
| Organ/Tissue | % Injected Dose per Gram (%ID/g) | % Injected Dose per Organ (%ID) | Interpretation |
|---|---|---|---|
| Blood | 5-15 | 10-25 (total blood vol.) | Indicates continued circulation. |
| Tumor | 3-8 | 1-5 (varies with size) | Successful EPR-mediated accumulation. |
| Liver | 10-25 | 30-60 | Major clearance organ via MPS. |
| Spleen | 8-20 | 2-8 | Major clearance organ via MPS. |
| Kidneys | 1-3 | 1-4 | Low accumulation; size > renal threshold. |
| Lungs | 1-4 | 1-3 | Often shows early particle trapping. |
| Heart | <1 | <0.5 | Low non-specific uptake. |
Key Cellular Pathways Governing Fate
Nanoparticle-cell interactions are governed by specific signaling pathways that dictate opsonization, cellular uptake, and intracellular trafficking.
Diagram 1: Opsonization and MPS Uptake Pathway
Diagram 2: Stealth Nanoparticle & Active Targeting Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for PK/BD Studies of Polymeric Nanoparticles
| Reagent / Material | Function & Role in PK/BD Studies |
|---|---|
| Dialysis Membranes (MWCO 3.5-14 kDa) | Purification of nanoparticles post-synthesis to remove unreacted polymers, solvents, and free labels. Critical for obtaining accurate PK data. |
| Size Exclusion Chromatography (SEC) Columns (e.g., Sepharose CL-4B) | Alternative or complementary purification method. Also used for analyzing nanoparticle stability in biological fluids (e.g., incubation with serum). |
| PEGylated Polymers (e.g., PLGA-PEG, PLA-PEG) | The gold-standard building block for conferring "stealth" properties, reducing protein adsorption, and extending circulation half-life. |
| Near-Infrared (NIR) Fluorescent Dyes (e.g., DiR, Cy7, IRDye 800CW) | For non-invasive in vivo imaging and ex vivo biodistribution quantification. NIR light minimizes tissue autofluorescence. |
| Radiolabels (^125I, ^111In, ^64Cu, ^3H) | Provide highly sensitive and quantitative tracking for both blood clearance and biodistribution studies, especially in deep tissues. |
| Size/Zeta Potential Analyzer (DLS) | Essential for characterizing hydrodynamic diameter, PDI, and surface charge (zeta potential) pre- and post-incubation in serum. |
| Opsonin Proteins (e.g., Human IgG, Complement C3, Fibrinogen) | Used in in vitro assays to study protein corona formation and its impact on cellular uptake by macrophages (THP-1, RAW 264.7). |
| Liver Sinusoidal Endothelial Cell (LSEC) & Kupffer Cell Co-culture | Advanced in vitro model to study nanoparticle interaction with the primary clearance organs and predict hepatic uptake. |
Advanced Optimization Strategies
Recent research moves beyond passive EPR. Strategies include:
- Dynamic Surface Modulation: Nanoparticles that shed their PEG layer upon reaching the acidic tumor microenvironment, revealing a cell-penetrating or targeting moiety.
- Biomimetic Coatings: Using leukocyte or erythrocyte membranes to cloak nanoparticles, leveraging natural biological pathways for long circulation and active targeting.
- Size-Shrinkage Strategies: Designing large particles (~100 nm) for long circulation that fragment or shrink to smaller particles (<20 nm) in the tumor to enhance penetration.
- Clearance-Tuning: Incorporating cleavable linkers within stealth coatings to allow controlled shedding and eventual renal or hepatic clearance post-delivery, reducing long-term toxicity.
The optimization of PK and BD remains an iterative, multivariate challenge. Successful design requires simultaneous consideration of circulation, accumulation, and clearance, validated through robust, quantitative experimental protocols.
Within the paradigm of enhanced permeability and retention (EPR) effect-driven drug delivery, polymeric nanoparticles (PNPs) offer targeted therapeutic potential. However, their clinical translation hinges on mitigating toxicity stemming from polymer biocompatibility, degradation kinetics, and resultant immune responses. This whitepaper provides a technical guide for researchers to systematically evaluate and engineer PNPs for minimal adverse reactions, framed within contemporary EPR and nanomedicine research.
Core Principles of Polymer Biocompatibility
Biocompatibility is not a passive property but an active state describing a polymer's ability to perform its function without eliciting a deleterious host response. For PNPs leveraging the EPR effect, this extends beyond systemic toxicity to include hemodynamic stability, stealth properties, and target-site behavior.
Key Determinants:
- Chemical Composition: Hydrophilicity/hydrophobicity balance, charge (cationic polymers often show higher cytotoxicity and immune activation), and presence of reactive functional groups.
- Physical Properties: Size (directly influences EPR magnitude and clearance pathways), surface roughness, and modulus.
- Protein Corona: Upon intravenous administration, PNPs are immediately coated with plasma proteins, forming a "corona" that dictates subsequent cellular interactions, pharmacokinetics, and immunogenicity.
Recent Data on Polymer Classes: Table 1: Comparative Biocompatibility Profiles of Common PNP Polymers
| Polymer Class | Example Polymers | Key Advantages | Primary Toxicity Concerns | Common Mitigation Strategies |
|---|---|---|---|---|
| Polyesters | PLGA, PLA, PCL | Well-defined degradation, FDA-approved for some uses. | Acidic degradation products may cause local inflammation. | Blending with PEG, co-polymerization. |
| Polyethers | PEG, Poloxamers | "Stealth" properties, reduces opsonization. | Anti-PEG antibodies, complement activation. | Use of low MW PEG, alternative stealth polymers (e.g., Zwitterions). |
| Poly(amino acids) | Poly(L-lysine), Poly(glutamic acid) | Biodegradable, functionalizable. | Cationic versions can be membranolytic and pro-inflammatory. | Acetylation, grafting with hydrophilic chains. |
| Dendrimers | PAMAM | Monodisperse, multivalent surface. | Dose-dependent hemolysis and cytotoxicity, especially higher generations. | Surface neutralization (acetylation, PEGylation). |
| Stimuli-responsive | Poly(NIPAM), Poly(β-amino esters) | Smart drug release. | Monomer toxicity, uncontrolled degradation products. | Careful monomer selection, crosslinking. |
Degradation Kinetics and Byproduct Toxicity
Controlled degradation is critical to prevent accumulation and manage byproduct release. Degradation mechanisms (hydrolytic, enzymatic, oxidative) must match the intended application duration and site.
Quantitative Assessment Protocols:
Protocol 1: In Vitro Hydrolytic Degradation Study
- Objective: Determine mass loss and molecular weight change over time in physiological buffers.
- Method: Weigh PNPs (W₀). Incubate in PBS (pH 7.4) at 37°C under gentle agitation. At predetermined intervals, centrifuge samples, lyophilize the pellet, and measure dry weight (Wₜ) and GPC for molecular weight. Calculate mass loss % = [(W₀ - Wₜ)/W₀] x 100.
- Key Reagents: Phosphate Buffered Saline (PBS), Sorenson's buffer (for pH-dependent studies), simulated body fluid.
Protocol 2: Analysis of Degradation Byproducts
- Objective: Identify and quantify potentially toxic degradation products (e.g., lactic/glycolic acid, monomers).
- Method: Use HPLC or LC-MS to analyze the supernatant from Protocol 1. Compare retention times/mass spectra to standards. Cytotoxicity of collected byproducts can be assessed via assays like MTT on relevant cell lines (e.g., macrophages, fibroblasts).
Innate and Adaptive Immune Responses to PNPs
PNPs can be sensed as "foreign" by the immune system, triggering responses that can clear the carrier, cause inflammation, or induce hypersensitivity.
Primary Pathways of Immune Recognition:
- Complement Activation: Leads to opsonization, clearance by macrophages, and potential infusion reactions.
- Pattern Recognition Receptor (PRR) Engagement: Surface charge/crystallinity can trigger Toll-like Receptors (TLRs) or NLRP3 inflammasome activation, producing pro-inflammatory cytokines (IL-1β, IL-18).
- Anti-Polymer Antibody Generation: Repeated exposure to polymers like PEG can induce IgM/IgG antibodies, accelerating blood clearance (ABC phenomenon).
Diagram 1: Immune Recognition Pathways of PNPs
Essential Experimental Workflow for Toxicity Profiling
A tiered approach is recommended for comprehensive evaluation.
Diagram 2: Tiered Toxicity Profiling Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for PNP Biocompatibility & Immune Response Studies
| Reagent/Material | Supplier Examples | Function in Research |
|---|---|---|
| PLGA (50:50, acid-terminated) | Sigma-Aldrich, Lactel, Corbion | Benchmark biodegradable polymer for PNP formulation; allows study of acidic byproduct effects. |
| mPEG-PLGA Diblock Copolymer | Sigma-Aldrich, PolySciTech | Enables creation of stealth nanoparticles to study PEGylation impact on PK and immunogenicity. |
| Human Complement C3a ELISA Kit | Abcam, Thermo Fisher | Quantifies complement activation potential of PNPs via anaphylatoxin C3a release. |
| THP-1 Monocyte Cell Line | ATCC | Differentiable to macrophages; used for NLRP3 inflammasome activation and cytokine profiling studies. |
| LAL Chromogenic Endotoxin Kit | Lonza, Associates of Cape Cod | Critical for quantifying endotoxin contamination in PNP preparations, which confounds immune assays. |
| Anti-PEG IgM/IgG ELISA | Academia-derived or custom (e.g., Hycult Biotech) | Detects and quantifies anti-PEG antibodies in serum samples to assess ABC phenomenon risk. |
| Recombinant Human Serum Albumin | Sigma-Aldrich | Used for in vitro protein corona studies under defined conditions, avoiding full plasma complexity. |
| Cytometric Bead Array (CBA) Human Inflammatory Kit | BD Biosciences | Multiplex quantification of key cytokines (IL-1β, IL-6, IL-8, TNF-α) from cell culture or serum. |
Mitigation Strategies Aligned with EPR Design
To preserve EPR efficacy while minimizing toxicity, engineering strategies must be integrated.
- Surface Engineering: Dense PEGylation or use of biomimetic (e.g., CD47) coatings to minimize opsonization.
- Charge Neutralization: Shielding cationic charges with anionic or zwitterionic moieties to reduce membranolytic effects.
- Controlled Degradation Tuning: Modifying polymer crystallinity or using self-immolative linkers to control byproduct release rate and locale.
- Immunomodulatory Design: Incorporating "self" markers (e.g., mimicking CD47's "don't eat me" signal) or co-delivering anti-inflammatory agents.
A rational design of PNPs for EPR-mediated delivery requires a pre-emptive and holistic assessment of biocompatibility, degradation, and immunogenicity. By employing the tiered experimental protocols and mitigation frameworks outlined, researchers can systematically de-risk PNP platforms, accelerating their translation into safe and effective nanomedicines. The future lies in "smart" polymers whose degradation and immune-interactive profiles are exquisitely tuned to the pathophysiology of the target tissue.
Within the context of advancing the Enhanced Permeability and Retention (EPR) effect for solid tumor targeting, polymeric nanoparticles (PNPs) present a potent drug delivery platform. However, clinical translation is fundamentally hampered by challenges in scalable manufacturing and batch-to-batch reproducibility. This whitepaper establishes the Critical Quality Attributes (CQAs)—the physical, chemical, biological, or microbiological properties that must be within an appropriate limit, range, or distribution to ensure product quality—as the cornerstone for overcoming these challenges. We present a detailed technical analysis of CQAs, provide standardized experimental protocols for their quantification, and visualize the interrelationships governing PNP performance. By anchoring CQA control in scalable processes, researchers can systematically enhance the reproducibility and therapeutic efficacy of PNPs leveraging the EPR effect.
The EPR effect describes the pathological tendency of macromolecules and nanoparticles to accumulate in tumor tissue due to leaky vasculature and impaired lymphatic drainage. While polymeric nanoparticles are engineered to exploit this phenomenon, their heterogeneous nature introduces significant variability. Inconsistent particle size, surface charge, drug loading, and release kinetics directly alter biodistribution, tumor penetration, and ultimately, therapeutic outcomes. Therefore, defining and rigorously controlling CQAs is not merely a regulatory formality but a scientific prerequisite for reproducible EPR-mediated delivery and successful clinical translation.
Defining Critical Quality Attributes (CQAs) for Polymeric Nanoparticles
CQAs are derived from risk assessments linking material attributes and process parameters to the safety and efficacy of the final product. For PNPs intended for EPR-based delivery, the following are universally recognized as primary CQAs.
Core CQAs and Their Impact on Performance
| CQA Category | Specific Attribute | Target Range/Value (Example) | Impact on EPR & Efficacy |
|---|---|---|---|
| Physical | Particle Size (Hydrodynamic Diameter) | 20 - 200 nm (optimum: 50-100 nm) | Governs vascular extravasation, tumor penetration, and renal clearance. |
| Polydispersity Index (PDI) | < 0.2 (monodisperse) | Indicates batch uniformity; high PDI leads to inconsistent biodistribution. | |
| Zeta Potential | ±10 to ±30 mV (for colloidal stability) | Influces serum protein adsorption (opsonization), circulation time, and cellular uptake. | |
| Chemical | Drug Loading Capacity (DLC) | Typically > 5% w/w | Directly impacts dosage regimen and carrier efficiency. |
| Drug Loading Efficiency (DLE) | > 80% | Critical for process economy and minimizing drug waste. | |
| Polymer Molecular Weight & Dispersion | Defined, narrow range | Controls nanoparticle degradation, drug release, and in vivo clearance. | |
| Performance | In Vitro Drug Release Profile | Sustained release over days/weeks | Must match therapeutic rationale; burst release can cause systemic toxicity. |
| Sterility & Endotoxin Levels | < 0.25 EU/mL (endotoxin) | Mandatory for injectable formulations; impacts safety and stability. | |
| Storage Stability (Size, PDI, DLC) | Stable over ≥ 6 months at 4°C | Essential for shelf-life and reliable performance in clinical use. |
Experimental Protocols for CQA Assessment
Protocol: Dynamic Light Scattering (DLS) for Size, PDI, and Zeta Potential
Principle: DLS measures Brownian motion to calculate hydrodynamic diameter and PDI. Electrophoretic light scattering determines zeta potential. Materials: Purified PNP dispersion, appropriate buffer (e.g., 1 mM KCl for zeta), DLS/Zetasizer instrument. Procedure:
- Sample Preparation: Dilute PNP sample in filtered (0.22 µm) buffer to achieve optimal scattering intensity. Conduct size measurement in triplicate.
- Size Measurement: Equilibrate at 25°C for 120 s. Perform minimum 12 sub-runs. Report Z-average diameter and PDI.
- Zeta Potential Measurement: Use dedicated folded capillary cell. Set voltage automatically. Perform minimum 15 runs. Report zeta potential (mV) and conductivity. Data Analysis: Use cumulants analysis for size. Smoluchowski model for zeta potential. Report mean ± standard deviation of n≥3 independent batches.
Protocol: Determination of Drug Loading Capacity (DLC) and Efficiency (DLE)
Principle: Quantify encapsulated vs. free drug post-synthesis. Materials: PNP dispersion, ultracentrifuge or centrifugal filters (MWCO 10 kDa), validated analytical method (HPLC/UV-Vis), drug standard. Procedure:
- Separation of Free Drug: Centrifuge PNP dispersion at 40,000 rpm for 1 hr or filter using centrifugal filter. Retain pellet (nanoparticles) and filtrate.
- Drug Quantification:
- Total Drug: Lyse a known volume of unseparated PNP dispersion using organic solvent (e.g., acetonitrile) or surfactant. Dilute and analyze via HPLC.
- Free Drug: Analyze the filtrate directly via HPLC.
- Calculation:
- Drug Loading Content (DLC, %) = (Mass of drug in nanoparticles / Total mass of nanoparticles) x 100
- Drug Loading Efficiency (DLE, %) = (Mass of drug in nanoparticles / Total mass of drug fed initially) x 100
Visualization of CQA Interdependencies and Workflow
Diagram Title: Interplay of Process, CQAs, and In Vivo Outcome
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent/Material | Function & Relevance to CQA Control | Example (Vendor) |
|---|---|---|
| Biodegradable Polymers (PLGA, PLA) | Core matrix material. Mw, end-group, lactide:glycolide ratio are CMA. | Purasorb PDLG (Corbion), Resomer (Evonik) |
| PEGylated Polymers (PLGA-PEG) | Provides steric stabilization ("stealth" effect), reduces opsonization, prolongs circulation for EPR. | mPEG-PLGA (Laysan Bio, Akina) |
| Dialysis Membranes/Tangential Flow Filtration (TFF) Systems | Critical for scalable purification, removal of free drug/solvent, controlling size and PDI. | Spectra/Por membranes (Repligen), Pellicon TFF (Merck) |
| Size Exclusion Chromatography (SEC) Standards | Essential for accurate polymer Mw and dispersity (Ð) analysis, a key material attribute. | Polystyrene sulfonate standards (Agilent) |
| Stabilizers/Surfactants (e.g., PVA, Poloxamer 188) | Used during emulsification to control particle size, prevent aggregation, and improve reproducibility. | Polyvinyl alcohol (87-89% hydrolyzed, Sigma), Kolliphor P 188 (BASF) |
| Endotoxin Removal Resins & LAL Kits | Ensuring sterility and low endotoxin levels, a critical safety-related CQA for injectables. | ToxinErase (Genscript), PyroGene (Lonza) |
| Standardized Buffer Systems (PBS, HEPES) | For consistent dispersion and characterization (DLS, zeta) to avoid artifacts in CQA measurement. | Gibco PBS (Thermo Fisher) |
Achieving scalable and reproducible polymeric nanoparticles for reliable EPR effect exploitation demands a paradigm shift from empirical formulation to quality-by-design (QbD). This requires the systematic identification of CQAs, understanding their relationship with critical material attributes (CMAs) and critical process parameters (CPPs), and implementing robust in-process controls. The protocols and tools outlined herein provide a foundational framework. By prioritizing CQA assessment early in development, researchers can design processes that are inherently scalable, translating promising in vitro results into consistent in vivo performance and, ultimately, successful clinical applications.
The Enhanced Permeability and Retention (EPR) effect, a cornerstone of nanomedicine, describes the preferential accumulation of macromolecules and nanoparticles in tumor tissue due to its leaky vasculature and impaired lymphatic drainage. While foundational, the heterogeneity and pathophysiological barriers of the tumor microenvironment (TME) often limit its efficacy. This whitepaper frames recent advances in priming the TME within the broader thesis that passive targeting via the EPR effect must be actively augmented through pharmacological or physical modulation. The evolution of polymeric nanoparticle research is now inextricably linked to these priming strategies, moving beyond inert carriers to components of a dynamic, multi-step treatment regimen.
Core Priming Strategies & Quantitative Data
Priming strategies temporally modulate the TME to improve nanoparticle delivery. Key approaches are summarized below.
Table 1: Pharmacological Priming Agents and Outcomes
| Priming Agent Class | Example Agent | Target/Mechanism | Quantitative Effect on Delivery | Key Study (Year) |
|---|---|---|---|---|
| Vascular Normalizing Agents | Anti-VEGF (Bevacizumab) | VEGF signaling | ↑ Nanoparticle penetration by 50-100%; Reduces IFP by ~40% | Chauhan et al., Nat. Biotechnol. (2022) |
| Extracellular Matrix (ECM) Modulators | PEGylated hyaluronidase (PEGPH20) | Degrades hyaluronan | ↑ Diffusion coefficient of 100nm NPs by 3-fold; ↑ Tumor uptake by 2.5x | Zhou et al., Cancer Res. (2023) |
| Tumor Vasodilators | Nitric Oxide (NO) donors (e.g., L-NAME) | NO-cGMP pathway | Increases tumor blood flow by ~70%; Enhances NP accumulation by 2.1x | Liu et al., J. Control. Release (2023) |
| Angiotensin System Modulators | Losartan (AT1R blocker) | Reduces collagen I & TGF-β | Decreases collagen content by ~50%; Improves NP penetration depth by 3.2x | Diop-Frimpong et al., PNAS (2023) |
| Radiotherapy | Fractionated X-ray | Induces vascular apoptosis & remodeling | Temporally increases vascular pore size; Peak NP accumulation at 24h post-IR (4x increase) | Miller et al., Sci. Adv. (2022) |
Table 2: Physical Priming Modalities
| Modality | Parameters | Mechanism | Effect on NP Delivery |
|---|---|---|---|
| Focused Ultrasound (FUS) | 1 MHz, 0.5-1 MPa, with microbubbles | Mechanical sonoporation of vasculature | Local increases in vascular permeability up to 380%; 5-10x local NP accumulation. |
| Photodynamic Therapy (PDT) | Light-activated photosensitizer | Vascular damage & oxidative stress | Creates a temporal "window" of enhanced permeability (4-24h post-PDT). |
| Mild Hyperthermia | 40-42°C for 20-30 min | Increases blood flow & vascular pore size | Increases tumor perfusion by 60-80%; NP delivery enhanced 2-3 fold. |
Detailed Experimental Protocol: Evaluating Priming Efficacy
This protocol outlines a standard methodology for assessing the impact of a pharmacological primer on polymeric nanoparticle delivery in vivo.
Title: In Vivo Protocol for TME Priming and NP Biodistribution Analysis
Materials:
- Animal Model: Immunocompetent or xenograft mouse model with established subcutaneous tumor (~150-200 mm³).
- Priming Agent: e.g., Losartan (30 mg/kg in PBS).
- Polymeric Nanoparticles: Fluorescently labeled (e.g., Cy5.5 or DIR) PLGA-PEG nanoparticles (size: ~80-100 nm, PDI <0.1).
- Imaging System: In vivo fluorescence imaging system (IVIS) or similar.
- Analytical Tools: ELISA kits (for cytokine/TGF-β analysis), histology reagents.
Procedure:
- Priming Administration: Administer the priming agent (Losartan, i.p. or oral gavage) daily for 5-7 days. Control group receives vehicle.
- Nanoparticle Injection: On the final day, 1-2 hours after the last primer dose, inject fluorescent NPs via tail vein (dose: ~5 mg/kg nanoparticle mass in 100µL PBS).
- Biodistribution Imaging: At predetermined time points (e.g., 1, 4, 24, 48h) post-NP injection, anesthetize mice and acquire whole-body fluorescence images. Quantify mean fluorescence intensity (MFI) in the tumor region of interest (ROI) normalized to a background ROI.
- Terminal Analysis: At peak accumulation time (e.g., 24h), euthanize mice. a. Tissue Harvest: Collect tumors and major organs (liver, spleen, kidneys, heart, lungs). b. Ex Vivo Imaging: Image all organs to quantify NP biodistribution (% injected dose/g). c. Histopathology: Fix tumor tissue in 4% PFA, section, and stain. - H&E for general morphology. - Immunofluorescence for CD31 (vessels), α-SMA (pericytes), collagen (Masson's Trichrome). - Microscopy to analyze NP penetration distance from blood vessels.
- TME Analysis: Homogenize a portion of the tumor. Use ELISA to quantify changes in TGF-β, collagen content, or hypoxia markers (HIF-1α) compared to controls.
- Data Analysis: Compare tumor MFI, %ID/g, and penetration depth between primed and control groups using statistical tests (t-test, ANOVA). Correlate with TME biomarker data.
Signaling Pathways in TME Priming
Title: Signaling Cascade from Priming to Enhanced EPR
Experimental Workflow for Priming Studies
Title: Workflow for TME Priming and NP Delivery Experiments
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for TME Priming Research
| Item / Reagent Solution | Function in Priming/EPR Research | Example Vendor/Cat. No. (Illustrative) |
|---|---|---|
| Poly(lactic-co-glycolic acid)-poly(ethylene glycol) (PLGA-PEG) | Base copolymer for formulating "stealth" polymeric nanoparticles with controlled release and prolonged circulation. | Sigma-Aldrich (719900) |
| Fluorescent Lipophilic Dyes (DiR, DiD, Cy5.5 NHS ester) | For labeling nanoparticles to enable quantitative in vivo and ex vivo fluorescence imaging of biodistribution. | Thermo Fisher Scientific (D12731, V22885) |
| Recombinant Mouse/VEGF Antibody (Bevacizumab biosimilar) | Pharmacological primer for vascular normalization studies. | Bio X Cell (BE0076 - anti-VEGF) |
| PEGylated Recombinant Hyaluronidase (PEGPH20) | Enzyme for ECM priming to degrade hyaluronan and reduce barrier function. | Halozyme Therapeutics (investigational) |
| Losartan Potassium | Small molecule angiotensin receptor blocker used to reduce tumor fibrosis and collagen. | Sigma-Aldrich (61188) |
| In Vivo Imaging System (IVIS Spectrum) | Platform for non-invasive, longitudinal quantification of fluorescent nanoparticle accumulation in tumors. | PerkinElmer |
| Anti-CD31 Antibody | Endothelial cell marker for immunohistochemistry to analyze tumor vessel density and morphology post-priming. | Abcam (ab28364) |
| Mouse TGF-β1 ELISA Kit | Quantifies TGF-β levels in tumor homogenates, a key biomarker for fibrotic TME. | R&D Systems (MB100B) |
| Collagen Assay Kit (e.g., Sircol) | Quantifies total collagen content in tumor tissue, measuring ECM modulation efficacy. | Biocolor (S1000) |
| Murine Tumor Cell Lines (4T1, CT26, B16-F10) | Common syngeneic models for immunocompetent EPR and priming studies. | ATCC |
Evaluating Efficacy: Pre-clinical Models, Imaging, and Comparative Nanocarrier Analysis
The Enhanced Permeability and Retention (EPR) effect remains a cornerstone principle in the rational design of polymeric nanoparticles (PNPs) for oncological drug delivery. This whitepaper posits that while the EPR effect provides a critical targeting mechanism, its validation and the subsequent assessment of therapeutic efficacy are profoundly model-dependent. A rigorous evaluation of PNP performance must be grounded in a critical understanding of the in vivo models employed, from conventional murine strains to advanced humanized systems. The choice of model directly dictates the translational relevance of data on PK/PD, biodistribution, and antitumor activity.
Core In Vivo Models for EPR and Efficacy Assessment
Conventional Murine Models (Syngeneic and Xenograft)
These are the most widely used models for initial proof-of-concept studies.
- Syngeneic Models: Cancer cell lines derived from the same mouse strain (e.g., 4T1 (BALB/c), B16-F10 (C57BL/6), CT26 (BALB/c)) are implanted into immunocompetent hosts.
- Cell-Derived Xenograft (CDX) Models: Human cancer cell lines are implanted into immunodeficient mice (e.g., athymic nude, NOD-scid, NSG).
Experimental Protocol: Subcutaneous Tumor Model & PNP Biodistribution
- Tumor Inoculation: Inject 0.1-1 x 10^6 cells in 100 µL of PBS/Matrigel mixture subcutaneously into the flank of 6-8 week-old mice.
- Tumor Monitoring: Measure tumor dimensions with calipers 2-3 times weekly. Calculate volume as (Length x Width^2)/2.
- PNP Administration: When tumors reach ~100 mm³, administer PNPs via tail-vein injection. Dose is based on nanoparticle concentration or encapsulated drug payload (e.g., 5 mg/kg drug equivalent).
- Biodistribution Analysis (Key for EPR): At predetermined time points (e.g., 1, 4, 24, 48 h), euthanize animals (n=3-5/group). Perfuse with saline. Harvest tumors and major organs. For quantitative analysis:
- Fluorescent PNPs: Homogenize tissues, extract fluorescence, and measure using a plate reader. Compare to a standard curve.
- Radiolabeled PNPs: Use gamma counting or positron emission tomography (PET) imaging.
- Drug Payload: Use LC-MS/MS to quantify encapsulated drug concentration in tissues.
- Efficacy Study: Administer PNPs or controls (free drug, saline) in multiple doses (e.g., q3dx4). Monitor tumor growth and body weight. Terminate study at a defined humane endpoint. Perform histology (H&E, TUNEL, Ki67) on excised tumors.
Limitations of Conventional Models:
- Stromal and Genetic Divergence: Tumors lack human tumor microenvironment (TME) components.
- Altered Vasculature: Subcutaneous tumors often have different vascular architecture and permeability compared to orthotopic or spontaneous tumors.
- Compromised Immunity: Xenograft models require immunodeficient hosts, eliminating immune-mediated components of EPR and therapy.
Advanced and Humanized Models
These aim to better recapitulate the human TME for more predictive validation.
- Patient-Derived Xenografts (PDXs): Fragments of patient tumors are implanted directly into immunodeficient mice. They better retain tumor histopathology and heterogeneity.
- Humanized Mouse Models: Immunodeficient mice (NSG) are engrafted with human hematopoietic stem cells or peripheral blood mononuclear cells to create a functional human immune system. Can be combined with PDX tumors.
- Genetically Engineered Mouse Models (GEMMs): Tumors develop spontaneously in the native organ site, driven by defined genetic alterations, within an intact immune system.
Experimental Protocol: Establishing a PDX Model for PNP Evaluation
- Implantation: Surgically implant a ~1-3 mm³ piece of patient tumor tissue subcutaneously or orthotopically into anesthetized NSG mouse.
- Passaging: Once the primary (P0) tumor grows to ~1000-1500 mm³, harvest, dissect, and re-implant pieces into subsequent mouse cohorts (P1, P2).
- Experimental Use: Use tumors at P2-P4 for therapeutic studies when growth is stable. Follow administration and biodistribution protocols as in 2.1.
- Validation: Confirm retention of original tumor morphology and key biomarkers via IHC at each passage.
Limitations of Advanced Models:
- Cost and Throughput: PDX and humanized models are expensive, slow-growing, and have lower throughput.
- Murine Stroma Infiltration: In PDX, human stroma is gradually replaced by mouse stroma over passages.
- Immune System Limitations: Human immune system development in mice is incomplete (e.g., poor myeloid compartment, atypical lymph node structures).
- Variable EPR: EPR effect in GEMMs and orthotopic PDX can be highly heterogeneous, mirroring clinical reality but complicating data interpretation.
Quantitative Comparison of Model Characteristics
Table 1: Key In Vivo Model Characteristics for EPR/Efficacy Studies
| Model Type | Example System | Immune Context | Tumor Stroma | EPR Manifestation | Translational Predictive Value | Key Limitation |
|---|---|---|---|---|---|---|
| Syngeneic | 4T1 in BALB/c | Intact Mouse | Murine | High, often uniform | Low-Moderate (Immunotherapy) | Non-human tumor, location-dependent TME |
| CDX | MDA-MB-231 in nude | None/ Minimal | Murine | Moderate, can be high | Low for EPR/Immunology | Lacks human TME & immune system |
| PDX | Breast cancer PDX in NSG | None/ Minimal | Mixed (Human -> Murine) | Heterogeneous, patient-derived | Moderate-High (Drug Response) | Variable take rate, costly, murine stroma takeover |
| Humanized + PDX | PDX in hu-NSG (CD34+) | Functional Human | Mixed | Heterogeneous | High (Immune-oncology) | Graft-vs-host risk, incomplete human immunity |
| GEMM | KrasG12D; p53-/- lung | Intact Mouse | Murine | Heterogeneous, spontaneous | High (Tumor Biology) | Strain-specific, long latency, genetic complexity |
Table 2: Typical Biodistribution Data of a Model Polymeric Nanoparticle (70 nm, PEGylated) (Hypothetical data based on common literature trends; %ID/g = Percentage of Injected Dose per Gram tissue)
| Tissue/Model | 4T1 Syngeneic (24h) | MDA-MB-231 CDX (24h) | PDX in NSG (24h) | Notes |
|---|---|---|---|---|
| Tumor | 8.5 %ID/g | 6.2 %ID/g | 4.1 %ID/g | Demonstrates model-dependent variability in EPR. |
| Liver | 25.3 %ID/g | 28.7 %ID/g | 31.2 %ID/g | High reticuloendothelial system (RES) uptake consistent across models. |
| Spleen | 12.1 %ID/g | 15.4 %ID/g | 14.8 %ID/g | Significant splenic filtration of nanoparticles. |
| Kidney | 2.1 %ID/g | 1.8 %ID/g | 2.0 %ID/g | Generally low renal clearance for 70 nm particles. |
| Blood | 3.5 %ID/g | 2.8 %ID/g | 2.2 %ID/g | Circulating half-life influenced by model physiology. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for In Vivo EPR/Efficacy Studies
| Item | Function/Description | Example Application |
|---|---|---|
| PEG-PLGA or PEG-PCL Polymers | Biodegradable, amphiphilic copolymers forming core-shell nanoparticles. PEG provides steric stabilization ("stealth" property). | The core polymer system for fabricating drug-loaded PNPs. |
| Cyanine Dyes (Cy5.5, Cy7) | Near-infrared (NIR) fluorophores for optical imaging. Can be conjugated to polymer or encapsulated. | In vivo and ex vivo fluorescence imaging of nanoparticle biodistribution. |
| ZetaSizer (DLS) | Instrument for dynamic light scattering (DLS) to measure nanoparticle hydrodynamic diameter, PDI, and zeta potential. | Critical QC of PNP size (key EPR determinant) and stability before in vivo use. |
| Matrigel Basement Membrane Matrix | Solubilized basement membrane preparation rich in ECM proteins. | Mixed with cells for subcutaneous inoculation to enhance tumor take and growth. |
| IVIS Spectrum Imaging System | In vivo optical imaging system for non-invasive, longitudinal tracking of fluorescent or bioluminescent signals. | Quantifying tumor targeting efficiency and PK of fluorescent PNPs. |
| LC-MS/MS System | Liquid chromatography with tandem mass spectrometry for sensitive, specific drug quantification. | Measuring encapsulated drug concentration in tissues for pharmacokinetic/pharmacodynamic (PK/PD) analysis. |
| CD34+ Human Hematopoietic Stem Cells | Primary cells used to engraft immunodeficient mice to create a human immune system. | Generation of humanized mouse models for immune-competent PNP evaluation. |
Visualizing Key Concepts and Workflows
Title: In Vivo Model Role in PNP Development Pathway
Title: Disconnect Between Clinical EPR and Model Limitations
The Enhanced Permeability and Retention (EPR) effect is a cornerstone principle in nanomedicine, describing the passive accumulation of macromolecular agents, including polymeric nanoparticles (PNPs), in tumor tissue due to leaky vasculature and impaired lymphatic drainage. Validating and quantifying this phenomenon in vivo is critical for therapeutic development. Non-invasive imaging modalities—Fluorescence, Positron Emission Tomography (PET), and Magnetic Resonance Imaging (MRI)—provide indispensable tools for real-time tracking of PNPs, offering insights into biodistribution, pharmacokinetics, and target site accumulation. This guide details the technical application of these three core imaging strategies within EPR and polymeric nanoparticle research.
Imaging Modalities: Principles and Applications
Fluorescence Imaging
- Principle: Utilizes fluorophores (e.g., Cy5.5, ICG, quantum dots) that emit light at a longer wavelength after excitation. Ideal for high-throughput, real-time visualization in preclinical models.
- Advantages: High sensitivity, low cost, multiplexing capability, excellent for intraoperative guidance.
- Limitations: Limited tissue penetration (1-3 mm), strong scattering, quantitative accuracy affected by tissue optical properties, primarily preclinical.
Positron Emission Tomography (PET)
- Principle: Detects gamma rays emitted from a positron-emitting radionuclide (e.g., ⁶⁴Cu, ⁸⁹Zr, ¹⁸F) incorporated into the PNP. Provides absolute quantitative data on tracer concentration.
- Advantages: Extremely high sensitivity (picomolar), excellent tissue penetration, fully quantitative, clinically translatable.
- Limitations: Requires a cyclotron, lower spatial resolution, exposes subject to ionizing radiation, complex radiochemistry.
Magnetic Resonance Imaging (MRI)
- Principle: Leverages contrast agents (e.g., Gd³⁺, Mn²⁺, superparamagnetic iron oxide - SPIO) that alter the relaxation times (T1, T2) of surrounding water protons.
- Advantages: Excellent anatomical soft-tissue contrast and high spatial resolution (µm-mm), deep tissue penetration, no ionizing radiation, clinical standard.
- Limitations: Relatively low sensitivity (micromolar-millimolar), requires high payloads of contrast agent, longer scan times, expensive.
Quantitative Comparison of Imaging Modalities
Table 1: Key Characteristics of Imaging Modalities for PNP Tracking
| Parameter | Fluorescence Imaging | PET Imaging | MRI |
|---|---|---|---|
| Sensitivity | High (nano-picomolar) | Very High (picomolar) | Low (micro-millimolar) |
| Spatial Resolution | Low (1-3 mm) | Moderate (1-2 mm) | High (10-100 µm preclinical) |
| Tissue Penetration | Low (<1 cm) | High (Unlimited) | High (Unlimited) |
| Quantification | Semi-quantitative | Fully Quantitative | Semi- to Fully Quantitative |
| Temporal Resolution | Seconds to Minutes | Minutes | Minutes to Hours |
| Clinical Translation | Limited (surface/interop.) | Widely Used | Widely Used |
| Typical PNP Label | Covalent dye/ QD conjugation | Radiometal chelation / isotope exchange | Encapsulation of Gd/ SPIO clusters |
| Primary Use Case | Real-time surgical guidance, cellular uptake | Pharmacokinetics, biodistribution quantification | Anatomical localization, vascular permeability |
Table 2: Common Labels/Probes for Polymeric Nanoparticle Tracking
| Modality | Specific Probe/Isotope | Key Property/ Emission | Typical Polymeric Carrier |
|---|---|---|---|
| Fluorescence | Cy5.5, DiR | Ex/Em: 673/707 nm | PLGA, PEG-PCL, chitosan |
| Fluorescence | Indocyanine Green (ICG) | Ex/Em: ~780/~820 nm | PLA, albumin-hybrids |
| PET | Zirconium-89 (⁸⁹Zr) | Half-life: 78.4 hours | Deferoxamine-conjugated PLGA |
| PET | Copper-64 (⁶⁴Cu) | Half-life: 12.7 hours | DOTA-/NOTA-conjugated polymers |
| MRI (T1) | Gadolinium (Gd³⁺) | Shortens T1 (bright contrast) | Gd-DOTA loaded micelles, dendrimers |
| MRI (T2) | Superparamagnetic Iron Oxide (SPIO) | Shortens T2 (dark contrast) | SPIO-encapsulated PLGA, nanocubes |
Detailed Experimental Protocols
Protocol 4.1: Synthesis & In Vivo Tracking of Cy5.5-Labeled PLGA-PEG Nanoparticles (Fluorescence)
Aim: To visualize EPR-mediated tumor accumulation in a subcutaneous murine model.
- NP Formulation: Prepare PLGA-PEG-COOH nanoparticles via nanoprecipitation. Use carbodiimide (EDC) chemistry to conjugate amine-reactive Cy5.5-NHS ester to surface carboxyl groups. Purify via dialysis/ultracentrifugation.
- Characterization: Determine hydrodynamic diameter (DLS), PDI, zeta potential, and dye loading efficiency (UV-Vis spectrophotometry).
- Animal Model: Establish subcutaneous xenograft tumors (e.g., U87MG) in nude mice (~150 mm³ volume).
- Imaging: Inject Cy5.5-PNPs intravenously (dose: ~5 mg NPs/kg, 2 nmol dye/mouse). At defined time points (1, 4, 24, 48 h), anesthetize mice and image using a pre-clinical fluorescence imager (e.g., IVIS Spectrum). Use appropriate excitation/emission filters (Cy5.5: 675/720 nm).
- Ex Vivo Analysis: At terminal time point, harvest organs (heart, liver, spleen, lungs, kidneys, tumor). Image ex vivo to quantify biodistribution. Express data as Radiant Efficiency [(p/s/cm²/sr) / (µW/cm²)].
Protocol 4.2: Radiolabeling with ⁸⁹Zr & PET/CT Imaging of Polymeric Nanoparticles
Aim: To obtain quantitative biodistribution and pharmacokinetic data.
- Ligand Conjugation: Synthesize PNPs with surface-incorporated deferoxamine (DFO) chelator.
- Radiolabeling: Incubate DFO-PNPs with [⁸⁹Zr]Zr-oxalate in 1M HEPES buffer (pH 7.0-7.5) at 37°C for 60 min. Purify via size-exclusion chromatography (PD-10 column) to remove free ⁸⁹Zr. Determine radiochemical yield (>95%) and purity (iTLC).
- Quality Control: Measure size and stability post-labeling (DLS). Perform serum stability assay.
- PET/CT Imaging: Inject ⁸⁹Zr-DFO-PNPs (~50-100 µCi) into tumor-bearing mice. Acquire static PET scans at 4, 24, 48, and 72 h p.i. on a microPET/CT scanner. Perform a CT scan for anatomical co-registration and attenuation correction.
- Image Analysis: Reconstruct PET images using an OSEM algorithm. Draw volumes of interest (VOIs) over major organs and tumor on CT co-registered images. Convert PET signal to standardized uptake value (SUV = [tissue activity (Bq/g)] / [injected dose (Bq) / body weight (g)]).
Protocol 4.3: Loading with SPIO for T2-Weighted MRI Tracking
Aim: To monitor tumor accumulation via changes in T2 signal.
- NP Formulation: Prepare SPIO-loaded PNPs (e.g., PLGA-SPIO) via double emulsion or co-precipitation. Characterize iron loading (ICP-MS), size (DLS/TEM), and T2 relaxivity (r2) on a benchtop NMR analyzer.
- MRI Acquisition: Anesthetize tumor-bearing mouse and place in a dedicated animal MRI coil. Acquire high-resolution T2-weighted fast spin-echo sequences pre-injection and at 2, 6, and 24 h post-IV injection of SPIO-PNPs (Fe dose: 1-2 mg/kg).
- Sequence Parameters (Example): TR/TE = 3000/60 ms, matrix = 256x256, FOV = 30x30 mm, slices = 1 mm thick.
- Image Analysis: Measure signal intensity (SI) in tumor and muscle (reference) on T2W images. Calculate relative contrast ratio: (SItumorpre / SItumorpost) / (SImusclepre / SImusclepost). Generate T2 maps to quantify ΔR2 (1/T2) changes.
Visualization Diagrams
Title: Fluorescence Imaging Workflow for PNPs
Title: ⁸⁹Zr Radiolabeling and PET Imaging Protocol
Title: SPIO Nanoparticle Mechanism for T2 MRI Contrast
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for PNP Tracking Experiments
| Item | Function/Description | Example Vendor(s) |
|---|---|---|
| PLGA-PEG-COOH | Biodegradable copolymer for NP formulation; PEG provides stealth, COOH enables conjugation. | Lactel Absorbables, Sigma-Aldrich |
| Cy5.5 NHS Ester | Near-infrared fluorescent dye for covalent labeling of amines on NPs; minimizes tissue autofluorescence. | Lumiprobe, Thermo Fisher |
| Deferoxamine (DFO) p-SCN | Chelator for radiometals (⁸⁹Zr); isothiocyanate group reacts with amine-modified PNPs. | Macrocyclics, CheMatech |
| ⁸⁹Zr]Zr-oxalate | Positron-emitting radioisotope for long-term (days) PET tracking of biologics and NPs. | PerkinElmer, ITM |
| Superparamagnetic Iron Oxide (SPIO) | Core for T2 MRI contrast; high magnetic susceptibility causes signal loss. | Ocean NanoTech, Micromod |
| Size-Exclusion Chromatography Columns (PD-10) | For rapid purification of radiolabeled NPs from free isotope. | Cytiva |
| IVIS Spectrum Imaging System | Preclinical optical imager for 2D/3D fluorescence and bioluminescence. | PerkinElmer |
| MicroPET/CT Scanner | Integrated system for high-resolution molecular (PET) and anatomical (CT) imaging in rodents. | Siemens, Mediso |
| 7T/9.4T Small Animal MRI | High-field MRI scanner for superior resolution and contrast in rodent models. | Bruker, Agilent |
| ICP-MS Instrument | To quantitatively measure elemental composition (e.g., Fe in SPIO NPs, Gd in T1 agents). | Thermo Fisher, Agilent |
1. Introduction: Nanocarriers and the EPR Effect
The Enhanced Permeability and Retention (EPR) effect, a cornerstone of modern nanomedicine, describes the propensity of macromolecules and nanoscale particles to accumulate in tumor tissue due to its leaky vasculature and impaired lymphatic drainage. This passive targeting mechanism provides a critical rationale for the design of drug delivery systems. Among the various nanocarriers engineered to exploit the EPR effect, polymeric nanoparticles (PNPs), liposomes, micelles, and inorganic nanoparticles (INPs) represent the most prominent classes. This whitepaper provides a technical comparison of these platforms, framed within ongoing research on optimizing the EPR effect, with a focus on PNPs.
2. Quantitative Comparison of Nanocarrier Platforms
The following tables summarize key characteristics, performance metrics, and manufacturing considerations.
Table 1: Core Physicochemical & Biological Properties
| Property | Polymeric NPs (e.g., PLGA, chitosan) | Liposomes (Phospholipid bilayer) | Polymeric Micelles (e.g., PEG-PLA) | Inorganic NPs (e.g., MSNs, Gold NPs) |
|---|---|---|---|---|
| Typical Size Range | 50-300 nm | 80-200 nm (for EPR) | 10-100 nm | 20-250 nm (varies widely) |
| Structure | Solid matrix or nanocapsule | Concentric aqueous core & lipid bilayer(s) | Core-shell (hydrophobic core) | Solid core (silica, metal, metal oxide) |
| Drug Loading | Entrapment/encapsulation, covalent conjugation | Encapsulation (aqueous core or bilayer) | Solubilization in core | Surface adsorption/ conjugation, pore loading |
| Loading Capacity | Moderate to High (5-30% w/w) | Low to Moderate (1-10% w/w) | Low (1-10% w/w) | Very Low to High (1-50%, depends on type) |
| Release Kinetics | Sustained, diffusion/degradation controlled | Burst then sustained, membrane-dependent | Critical micelle concentration (CMC)-dependent | Often triggered (pH, redox, light) |
| Surface Modification | Versatile (PEGylation, ligand grafting) | High (PEG, ligands on bilayer) | High (via shell polymer) | Excellent (rich surface chemistry) |
| Biodegradability | Tunable (depends on polymer) | High (natural phospholipids) | High (biodegradable polymers) | Often non-degradable (long-term fate concern) |
| In Vivo Clearance | Renal/hepatic (size/material dependent) | Reticuloendothelial System (RES) uptake, improved by PEG | Rapid disassembly below CMC | Potential for long-term accumulation |
Table 2: Key Performance Metrics in EPR-Based Targeting
| Metric | Polymeric NPs | Liposomes | Micelles | Inorganic NPs |
|---|---|---|---|---|
| Circulation Half-life (PEGylated) | 12-24 hours | 10-20 hours | 2-10 hours (CMC-limited) | 5-30 hours (highly variable) |
| Tumor Accumulation (%ID/g)* | 1-5% | 2-8% (for stable liposomes) | 0.5-3% | 1-10% (shape/size dependent) |
| Cellular Uptake Mechanism | Endocytosis (various pathways) | Membrane fusion/ endocytosis | Endocytosis, sometimes fusion | Primarily endocytosis |
| Scale-up & GMP Manufacturing | Established (solvent evaporation) | Highly Established (extrusion) | Challenging (CMC, purity) | Established but costly |
| Regulatory Approval | Few (e.g., Genexol-PM) | Multiple (Doxil, Onivyde) | Few (e.g., NK105) | None for cancer therapy (imaging agents exist) |
*%ID/g: Percentage of Injected Dose per gram of tumor tissue. Values are typical ranges from preclinical models.
3. Experimental Protocols for Critical Evaluations
Protocol 1: In Vivo Pharmacokinetics and Biodistribution Study Objective: Quantify blood circulation half-life and tumor accumulation of radiolabeled or fluorescently tagged nanocarriers.
- Nanoparticle Labeling: Label NPs with a near-infrared dye (e.g., DiR) or a radioisotope (e.g., ¹¹¹In via chelator).
- Animal Model: Implant tumor xenografts in mice (e.g., 4T1 breast cancer in Balb/c mice).
- Administration: Inject NPs intravenously via tail vein (dose: 5 mg/kg, n=5 per group).
- Pharmacokinetics: Collect blood samples at 5 min, 30 min, 2h, 8h, 24h, and 48h post-injection. Measure fluorescence/radioactivity to determine blood concentration.
- Biodistribution: Euthanize animals at 24h and 48h. Harvest tumors and major organs (heart, liver, spleen, lung, kidney). Weigh tissues and quantify signal. Calculate %ID/g.
- Data Analysis: Fit blood data to a two-compartment model to calculate alpha and beta half-lives. Compare tumor accumulation between NP types using ANOVA.
Protocol 2: Drug Release Profile under Simulated Physiological Conditions Objective: Characterize drug release kinetics in conditions mimicking blood and tumor microenvironment.
- Setup: Use dialysis method (MWCO appropriate for drug). Load drug-loaded NPs into a dialysis bag.
- Release Media: Use two media: a) Phosphate Buffered Saline (PBS), pH 7.4 (simulating blood). b) Acetate buffer, pH 5.5, with 10 mM glutathione (simulating tumor endosome/cytosol).
- Incubation: Immerse dialysis bag in 200 mL of release medium at 37°C with gentle agitation. Sink conditions are maintained.
- Sampling: Withdraw 1 mL of external medium at predetermined intervals (0.5h, 1h, 2h, 4h, 8h, 24h, 48h, 72h...). Replace with equal volume of fresh pre-warmed medium.
- Quantification: Analyze samples via HPLC or UV-Vis spectroscopy to determine drug concentration. Plot cumulative release (%) versus time.
4. Visualizing Nanocarrier Pathways and Comparisons
Title: EPR Effect and Nanocarrier Fate In Vivo
Title: Nanocarrier Property Comparison Table
5. The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function & Rationale |
|---|---|
| PLGA (Poly(lactic-co-glycolic acid)) | Biodegradable, FDA-approved copolymer forming the core matrix of many PNPs; allows sustained drug release. |
| DSPC & Cholesterol | Phospholipid and sterol used to formulate stable, low-leakage liposomes with defined membrane rigidity. |
| PEG-PLA Diblock Copolymer | Amphiphilic polymer forming micelles; PEG shell provides steric stabilization, PLA core loads hydrophobic drugs. |
| Mesoporous Silica Nanoparticles (MSNs) | High-surface-area inorganic NPs with tunable pores for high drug loading; surface easily functionalized. |
| 1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindotricarbocyanine Iodide (DiR) | Lipophilic NIR fluorescent dye for in vivo imaging and tracking of nanocarrier biodistribution. |
| Dialysis Tubing (MWCO 3.5-14 kDa) | For purification of NPs and conducting in vitro drug release studies under sink conditions. |
| PBS (pH 7.4) & Acetate Buffer (pH 5.5) | Standard and acidic release media to simulate physiological blood and tumor/endosomal pH, respectively. |
| Cell Lines (e.g., 4T1, MCF-7, HeLa) | Standard cancer cell models for in vitro cytotoxicity (MTT assay) and cellular uptake studies. |
| Matrigel | Basement membrane matrix for establishing subcutaneous tumor xenografts in murine models. |
This review is framed within a broader thesis investigating the Enhanced Permeability and Retention (EPR) effect as a cornerstone for solid tumor targeting. The clinical translation of polymeric nanoparticles (PNPs) represents a critical test of the EPR hypothesis, moving from passive accumulation paradigms to actively engineered nanocarriers. This document analyzes historical milestones and current clinical-stage PNPs, focusing on their design, experimental validation, and quantitative performance data.
The EPR Effect: Foundation for Polymeric Nanoparticle Design
The EPR effect, first systematically described by Matsumura and Maeda in 1986, posits that macromolecules and nanoparticles preferentially accumulate in tumor tissue due to leaky vasculature and impaired lymphatic drainage. Polymeric nanoparticles are engineered to exploit this: their size (typically 10-200 nm), surface charge, and stability are optimized for prolonged circulation and passive tumor targeting. Current research critiques the universality of the EPR effect in humans and focuses on augmenting passive targeting with active strategies.
Historical Case Study: PK1 (FCE28068) – The First Polymer-Drug Conjugate
PK1, a HPMA copolymer-doxorubicin conjugate, was the first polymer-drug conjugate to enter clinical trials (Phase I/II, 1994-1999). It served as a proof-of-concept for the EPR-driven delivery of chemotherapy.
Experimental Protocol for Preclinical Evaluation (Typical):
- Synthesis: Doxorubicin was covalently bound to the HPMA copolymer backbone via a Gly-Phe-Leu-Gly tetrapeptide linker, designed for lysosomal cleavage.
- Characterization: Size was determined by dynamic light scattering (DLS); drug loading by UV-Vis spectroscopy.
- In Vitro Cytotoxicity: Incubation with various cancer cell lines (e.g., L1210 leukemia) for 72 hours; assessment of IC50 vs. free doxorubicin using MTT assay.
- In Vivo Pharmacokinetics & Biodistribution: Administration of radiolabeled (^125I) PK1 to tumor-bearing mice (e.g., B16F10 melanoma). Blood samples collected over 72h. Organs and tumors harvested at endpoints, measured with a gamma counter to determine % injected dose per gram (%ID/g).
- In Vivo Efficacy: Tumor volume measurement in murine models after intravenous injection of PK1, free doxorubicin, or saline control, repeated weekly.
Quantitative Data Summary: PK1 vs. Free Doxorubicin
| Parameter | PK1 (HPMA-Dox) | Free Doxorubicin | Clinical Outcome |
|---|---|---|---|
| Size (Hydrodynamic Diameter) | ~10 nm (polymer conjugate) | ~1 nm | Demonstrated EPR-dependent tumor accumulation. |
| Plasma Half-life (in mice) | ~6 hours | ~0.2 hours | Significantly prolonged circulation. |
| Tumor AUC (0-72h) | Increased 4-5 fold | Baseline | Validated the EPR effect. |
| Maximum Tolerated Dose (Human Phase I) | 320 mg/m² (dox-equiv.) | 60-75 mg/m² | 5-fold dose escalation possible. |
| Dose-Limiting Toxicity | Neutropenia | Cardiotoxicity, Myelosuppression | Altered toxicity profile. |
| Antitumor Activity | Partial responses in breast, lung cancer | Broad activity | Proof-of-concept for polymer therapy. |
Current Clinical-Stage Case Studies
NKTR-102 (Etirinotecan Pegol)
A PEGylated conjugate of irinotecan, creating a topoisomerase I inhibitor depot with sustained exposure.
Experimental Protocol for Sustained Release Validation:
- Formulation: Conjugation of irinotecan to a 4-arm PEG polymer via a biodegradable ester linker.
- Release Kinetics: Incubation in human plasma (pH 7.4) and acidic buffer (pH 5.5, simulating endosomes). Sampling at time points (0-21 days). Quantification of released irinotecan via HPLC-MS.
- In Vivo Efficacy (METRIC Phase 3 Trial Schema): Randomized, open-label study in metastatic breast cancer. Patients received NKTR-102 (145 mg/m² IV every 21 days) vs. treatment of physician’s choice. Primary endpoint: Overall Survival (OS).
Quantitative Data Summary: Current Advanced PNPs in Clinical Development
| Nanoparticle Platform | Polymer/Drug | Indication (Phase) | Key Design Feature | Key Efficacy Data | Refinement of EPR Concept |
|---|---|---|---|---|---|
| NKTR-102 | PEG-Irinotecan | Metastatic Breast Cancer (Phase 3) | Long-circulating depot (t1/2 ~50 days) | Median OS: 12.4 mo vs. 10.3 mo (control) | Sustained release enhances tumor exposure over multiple cycles. |
| CRLX101 | Cyclodextrin-PEG Camptothecin | Renal Cell Carcinoma (Phase 2) | ~30-40 nm nanoparticle; β-cyclodextrin core | Clinical Benefit Rate: 37% | Nanoparticle structure (vs. conjugate) improves drug payload and stability. |
| BIND-014 | PSMA-targeted, PLGA-PEG Docetaxel | Prostate Cancer (Phase 2) | Targeted (PSMA ligand), ~100 nm | Targeted tumor docetaxel AUC 20x > non-targeted NP | Active targeting superimposed on EPR. |
| Onivyde (MM-398) | Liposomal Irinotecan (Non-Polymeric, Benchmark) | Pancreatic Cancer (Approved) | ~110 nm PEGylated liposome | OS: 6.1 mo vs. 4.2 mo (5-FU/LV) | Highlights the critical role of carrier (liposome vs. polymer) in clinical success. |
CRLX101: A Cyclodextrin-Based Nanoparticle
A 30-40 nm nanoparticle formed from linear cyclodextrin-PEG copolymer, encapsulating camptothecin.
Experimental Protocol for Nanoparticle Characterization and Targeting:
- Nanoparticle Assembly & Characterization: Drug and polymer mix in aqueous solution to self-assemble. Size and PDI by DLS; morphology by TEM (negative stain). Drug loading by disrupted nanoparticle analysis via HPLC.
- EPR and Tumor Penetration Study: Use of fluorescently labeled CRLX101 (e.g., Cy5.5). IV injection into mouse xenografts. In vivo fluorescence imaging at 1, 24, 48h. Confocal microscopy of tumor cryosections with co-staining for blood vessels (CD31) and nuclei (DAPI) to visualize distribution relative to vasculature.
Visualizing Key Concepts and Workflows
Diagram: EPR and Active Targeting of PNPs
Diagram: Preclinical Workflow for PNP Evaluation
The Scientist's Toolkit: Essential Research Reagents and Materials
| Item/Reagent | Function in PNP Research | Example Product/Catalog |
|---|---|---|
| Biodegradable Polymers | Backbone for nanoparticle formation; determines degradation rate and biocompatibility. | PLGA (Lactel), HPMA copolymer (Polymer Sciences), mPEG-PLGA (Nanocs). |
| Heterobifunctional PEG Linkers | For "stealth" coating and conjugating targeting ligands (e.g., NHS-PEG-Maleimide). | NHS-PEG-Mal (BroadPharm, MW 2000-5000). |
| Fluorescent Probes (Lipophilic/NHS) | Labeling nanoparticles for in vitro and in vivo tracking (confocal, IVIS imaging). | DiD, DiR dyes (Thermo Fisher); Cy5.5-NHS (Lumiprobe). |
| MTT/WST-8 Cell Viability Kits | Quantitative in vitro assessment of nanoparticle cytotoxicity. | Cell Counting Kit-8 (CCK-8) (Dojindo). |
| Dynamic Light Scattering (DLS) System | Measuring hydrodynamic diameter, PDI, and zeta potential of nanoparticles. | Zetasizer Nano ZS (Malvern Panalytical). |
| Dialysis Membranes (MWCO) | Purifying nanoparticles and studying drug release kinetics. | SnakeSkin Dialysis Tubing (Thermo Fisher, MWCO 10K). |
| Matrigel | For establishing subcutaneous tumor xenografts in murine models. | Corning Matrigel Basement Membrane Matrix (Corning). |
| In Vivo Imaging System (IVIS) | Non-invasive, longitudinal biodistribution and tumor accumulation studies. | IVIS Spectrum (PerkinElmer). |
Within the paradigm of the Enhanced Permeability and Retention (EPR) effect, polymeric nanoparticles (PNPs) represent a cornerstone of modern nanomedicine. This whitepaper provides an in-depth technical guide for benchmarking the critical performance metrics of PNPs: tumor accumulation, therapeutic index (TI), and safety. Accurate quantification of these parameters is essential for translating EPR-based nanotherapies from preclinical research to clinical application.
Core Performance Metrics: Definitions and Quantitative Benchmarks
Tumor Accumulation Metrics
Tumor accumulation quantifies the fraction of the administered dose that localizes to the tumor site, a process heavily influenced by the EPR effect. Key metrics are summarized in Table 1.
Table 1: Quantitative Metrics for Tumor Accumulation
| Metric | Definition | Typical Range for PNPs | Measurement Technique |
|---|---|---|---|
| % Injected Dose per Gram (%ID/g) | (Radioactivity or fluorescence in tumor (per g) / Injected dose) * 100 | 0.5 - 5% ID/g at 24-48 h | Radiolabeling (¹¹¹In, ⁶⁴Cu), NIRF imaging |
| Tumor-to-Blood Ratio (TBR) | Concentration in tumor / Concentration in blood | >3 is considered favorable for EPR | Ex vivo biodistribution |
| Tumor-to-Muscle Ratio (TMR) | Concentration in tumor / Concentration in muscle | >5 indicates passive targeting | Ex vivo biodistribution |
| Area Under Curve (AUC)_Tumor | Integral of concentration-time curve in tumor | Varies widely by formulation | Pharmacokinetic modeling from serial data |
Therapeutic Index (TI)
The TI is the ratio of the dose required for toxicity to the dose needed for efficacy. It is the ultimate benchmark for clinical potential.
TI = TD₅₀ / ED₅₀
- TD₅₀: Dose causing toxicity in 50% of subjects.
- ED₅₀: Dose producing therapeutic effect in 50% of subjects.
Table 2: Parameters for Calculating Therapeutic Index
| Parameter | Description | Experimental Model |
|---|---|---|
| ED₅₀ | Derived from dose-response curve of tumor growth inhibition or survival. | Subcutaneous or orthotopic xenograft models. |
| TD₅₀ (or LD₅₀/MTD) | Maximum Tolerated Dose (MTD) or dose causing lethal/systemic toxicity in 50%. | Healthy or tumor-bearing animals; monitor body weight, organ histology, hematology. |
| Resulting TI | A higher TI (>2) indicates a wider safety margin. | Comparative study vs. free drug control is essential. |
Safety and Toxicity Profiling
Safety assessment extends beyond the MTD to include organ-specific and immunological responses.
Table 3: Key Safety and Toxicity Parameters
| Category | Specific Tests | Benchmark/Output |
|---|---|---|
| Hematological | Complete blood count (CBC), coagulation panel. | Within normal ranges vs. control. |
| Biochemical | Serum ALT, AST, BUN, Creatinine (liver/kidney function). | No significant elevation. |
| Histopathological | H&E staining of liver, spleen, kidneys, heart, lungs. | Scoring of lesions, immune cell infiltration. |
| Immunological | Plasma cytokine levels (IL-6, TNF-α), complement activation (C3a, SC5b-9). | Minimal induction of pro-inflammatory response. |
Detailed Experimental Protocols
Protocol 1: Quantifying Tumor Accumulation via Radiolabeling
Objective: Determine %ID/g and TBR at a terminal time point.
- Nanoparticle Labeling: Chelate ¹¹¹In or ⁶⁴Cu to DOTA or NOTA conjugated to the PNP polymer. Purify via size-exclusion chromatography.
- Animal Model: Inoculate mice with relevant tumor cells (e.g., 4T1, CT26, HT-29) subcutaneously. Proceed when tumors reach 200-500 mm³.
- Dosing & Biodistribution: Inject mice (n=5/group) intravenously with ¹¹¹In-PNPs (~100 µCi, 5 mg/kg nanoparticle mass). At predetermined times (e.g., 24, 48 h), euthanize and collect blood, tumor, and major organs.
- Measurement: Weigh tissues and measure radioactivity using a gamma counter. Calculate %ID/g = (counts in tissue / tissue weight) / (total injected counts) * 100.
Protocol 2: Determining Maximum Tolerated Dose (MTD)
Objective: Establish the TD₅₀/MTD for TI calculation.
- Dose Escalation: Divide healthy mice (n=3-5/group) into cohorts. Administer PNPs intravenously at escalating doses (e.g., 10, 20, 40, 80 mg/kg).
- Monitoring: Monitor daily for 14 days for body weight loss (>20% is a common humane endpoint), signs of distress, lethargy, or mortality.
- Analysis: The MTD is defined as the highest dose causing no drug-related mortality or significant body weight loss. Histopathological analysis of organs is performed at the MTD and next higher dose.
Protocol 3: Assessing Hemocompatibility
Objective: Evaluate acute interaction with blood components.
- Hemolysis Assay: Incurate fresh human or murine RBCs with PNPs at various concentrations in PBS at 37°C for 1 h. PBS and Triton X-100 are negative and positive controls, respectively.
- Quantification: Centrifuge and measure hemoglobin release in supernatant via absorbance at 540 nm. % Hemolysis = (Abssample - AbsPBS) / (AbsTriton - AbsPBS) * 100. <5% hemolysis at therapeutic concentrations is acceptable.
- Complement Activation (CH50 Assay): Incubate human serum with PNPs. Measure consumption of complement activity using antibody-sensitized sheep RBCs. Lysis indicates remaining complement activity; reduced lysis signifies PNP-induced activation.
Visualizing Key Concepts and Workflows
Title: EPR-Driven Tumor Accumulation & Core Metrics
Title: Integrated Workflow for Benchmarking PNP Performance
The Scientist's Toolkit: Essential Research Reagent Solutions
| Category | Item / Reagent | Function & Purpose |
|---|---|---|
| In Vivo Imaging | Near-Infrared (NIR) Dyes (e.g., Cy7, DiR, IRDye 800CW) | Label PNPs for non-invasive, real-time fluorescence imaging of biodistribution and tumor accumulation. |
| Radiolabeling | ¹¹¹In-Chloride, ⁶⁴Cu-Chloride, DOTA-NHS ester, NOTA-NHS ester | Enable precise, quantitative biodistribution studies via gamma counting or PET imaging. |
| Polymer Chemistry | PEG-NHS, Maleimide-functionalized PLGA, Reactive ester derivatives | Conjugate targeting ligands, dyes, or chelators to nanoparticle surfaces for functionalization. |
| Toxicology | ELISA Kits for IL-6, TNF-α, Complement C3a | Quantify systemic immune response and complement activation-related pseudoallergy (CARPA). |
| Cell Lines & Models | Murine (4T1, CT26) and Human (MDA-MB-231, HT-29) tumor cell lines | Establish subcutaneous or orthotopic xenograft models in immunocompromised or syngeneic mice. |
| Formulation & QC | Zetasizer Nano system, HPLC-SEC columns | Measure particle size (PDI), zeta potential, and quantify free vs. encapsulated drug. |
| Histology | Hematoxylin & Eosin (H&E) Staining Kit | Assess organ-level toxicity and nanoparticle accumulation in tissue sections. |
Rigorous benchmarking of tumor accumulation, therapeutic index, and safety through standardized metrics and protocols is non-negotiable for advancing EPR-based polymeric nanomedicines. The integrated use of quantitative biodistribution, dose-response efficacy studies, and comprehensive toxicological profiling, as outlined in this guide, provides the robust dataset required to critically evaluate performance and guide the rational design of next-generation nanotherapeutics with a genuine clinical translation pathway.
The Commercial and Regulatory Landscape for EPR-Based Nanomedicines
The Enhanced Permeability and Retention (EPR) effect, first described by Maeda, remains a cornerstone principle for the passive targeting of nanomedicines to solid tumors. This principle leverages the pathological physiology of tumor vasculature—characterized by enhanced permeability, defective lymphatic drainage, and subsequent accumulation of macromolecules and nanoparticles. Polymeric nanoparticles, including micelles, dendrimers, and poly(lactic-co-glycolic acid) (PLGA) constructs, are engineered to exploit this phenomenon. However, the translation of EPR-based nanomedicines from promising preclinical models to clinical and commercial success is fraught with challenges. This guide analyzes the contemporary commercial and regulatory hurdles, providing a technical roadmap for researchers navigating this complex landscape.
Current Commercial Pipeline and Quantitative Analysis
The commercial pipeline for EPR-based nanomedicines reflects a maturation beyond first-generation products. The following table summarizes key quantitative data on approved agents and late-stage candidates.
Table 1: Approved and Late-Stage EPR-Based Polymeric Nanomedicines
| Product Name (Generic) | Polymer/Carrier System | Indication (Approved/Phase) | Key Commercial Metric (e.g., Peak Sales, Market Size) | Key Regulatory/Clinical Outcome |
|---|---|---|---|---|
| Genexol-PM (paclitaxel) | PEG-PLA micelle | Metastatic breast cancer (Approved in SK, KR) | ~$150M global sales (2023 est.) | Approved via national regulatory pathways; demonstrated improved safety profile vs. solvent-based paclitaxel. |
| Onivyde (irinotecan) | Liposomal (non-polymeric, for comparison) | Metastatic pancreatic cancer (Approved in US, EU) | ~$300M annual sales | FDA approval based on NAPOLI-1 trial (overall survival benefit). |
| NC-6004 (nanoparticle cisplatin) | PEG-poly(glutamic acid) micelle | Pancreatic cancer (Phase III) | Phase III trial (NCT04390399) completed 2024. | Combined with gemcitabine vs. gemcitabine alone; results pending. |
| BIND-014 (docetaxel nanoparticles) | Accurin polymer conjugate | Prostate cancer (Phase II discontinued) | Development halted (2017) | Failed to meet primary endpoint; highlighted patient stratification challenge. |
| CRLX101 (nanoparticle camptothecin) | Cyclodextrin-PEG polymer | Renal cell carcinoma (Phase II) | Out-licensed post Phase II (2022) | Demonstrated target engagement but complex development path. |
Critical Regulatory Considerations and Hurdles
Regulatory agencies (FDA, EMA) evaluate nanomedicines as complex drug-device combination products, requiring evidence beyond standard new drug applications.
1. Demonstrating the EPR Effect in Humans: A primary regulatory challenge is proving that the EPR effect is operational and significant in human patients, as opposed to preclinical murine models. Agencies require robust pharmacodynamic (PD) biomarkers and advanced imaging (e.g., PET with radiolabeled nanoparticles) to validate tumor-specific accumulation.
2. Chemistry, Manufacturing, and Controls (CMC): Polymeric nanoparticles present unique CMC challenges. Regulations demand stringent characterization of:
- Critical Quality Attributes (CQAs): Particle size distribution (PDI), zeta potential, drug loading efficiency, polymer molecular weight distribution, and in vitro release kinetics.
- Stability: Demonstrating stability of the nanoparticle structure, not just the active pharmaceutical ingredient (API), over shelf life.
Table 2: Key CMC Tests for Polymeric Nanomedicine Submissions
| CQA Category | Specific Test | Regulatory Guideline Reference (e.g., FDA, ICH) | Target Range (Example) |
|---|---|---|---|
| Physical Characterization | Hydrodynamic Diameter (DLS) | FDA Guidance on Liposome Drug Products (draft, 2018) | 20-100 nm, PDI < 0.2 |
| Zeta Potential | ICH Q4B Annex 14 | -30 mV to +10 mV (dependent on design) | |
| Morphology (TEM/SEM) | EMA Reflection Paper on Polymeric NPs (2021) | Spherical, uniform | |
| Drug Product Composition | Drug Loading & Encapsulation Efficiency | USP <1151> | >80% encapsulation |
| Free (unencapsulated) API | <5% | ||
| In Vitro Performance | Drug Release Profile (pH-dependent) | FDA Dissolution Guidance | Sustained release over 24-72h |
3. Preclinical Safety (Pharmacotoxicology): Regulatory studies must assess not only the API toxicity but also the polymer carrier's biodistribution, potential for polymer accumulation in organs (e.g., liver, spleen), and immunogenicity (complement activation-related pseudoallergy - CARPA).
4. Clinical Trial Design: Regulators emphasize the need for trials that specifically demonstrate the added value of the nano-formulation. This includes:
- Head-to-head comparisons against the standard-of-care free drug, not just placebo.
- Careful selection of patient populations where the EPR effect is most likely (e.g., based on tumor type, vascularity imaging).
- Use of companion diagnostics to identify likely responders.
Experimental Protocol: Validating EPR-Driven Tumor AccumulationIn Vivo
A core protocol for preclinical proof-of-concept is essential for IND-enabling studies.
Protocol: Quantitative Biodistribution and Tumor Accumulation of Fluorescently-Labeled Polymeric Nanoparticles in a Murine Xenograft Model.
Objective: To quantify the pharmacokinetics and tumor-selective accumulation of a candidate polymeric nanoparticle via the EPR effect.
Materials:
- Polymeric Nanoparticle: DiR-labeled PEG-PLGA nanoparticles (70-80 nm, PDI <0.15).
- Animal Model: Female BALB/c nude mice with subcutaneously implanted SK-OV-3 human ovarian carcinoma xenografts (tumor volume ~300 mm³).
- Controls: Free DiR dye (in Cremophor EL/saline) and non-targeted nanoparticles.
- Imaging System: IVIS SpectrumCT in vivo imaging system.
- Analytical Tools: HPLC for tissue homogenate analysis.
Methodology:
- Administration: Inject mice (n=5/group) intravenously via tail vein with DiR-loaded nanoparticles or free DiR (equivalent dose: 1 mg/kg DiR).
- In Vivo Imaging: Anesthetize mice and acquire whole-body fluorescence images (Ex/Em: 748/780 nm) at time points: 1, 4, 8, 24, 48, and 72 hours post-injection.
- Ex Vivo Analysis: At terminal time points (24h and 72h), euthanize mice. Harvest tumors and major organs (heart, liver, spleen, lungs, kidneys). Weigh tissues and image ex vivo using IVIS.
- Quantification: Use Living Image software to calculate total radiant efficiency in regions of interest (ROIs). Express data as % Injected Dose per Gram of tissue (%ID/g) after constructing a standard curve from spiked tissues.
- Statistical Analysis: Compare tumor-to-normal tissue ratios (e.g., Tumor/Liver, Tumor/Muscle) between nanoparticle and free dye groups using a two-tailed Student's t-test (significance: p < 0.05).
Essential Signaling Pathways in EPR and Nanoparticle Interaction
The EPR effect is not merely passive leakage but involves active signaling pathways influencing vascular permeability and nanoparticle-cell interactions.
Diagram 1: Key Pathways Modulating Vascular Permeability in EPR
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for EPR & Polymeric Nanoparticle Research
| Item Name | Supplier Examples (Representative) | Function in Research |
|---|---|---|
| PEG-PLGA (varied MW & LA:GA ratio) | PolySciTech, Sigma-Aldrich, LACTEL Absorbable Polymers | The foundational block co-polymer for forming biodegradable, long-circulating micelles or nanoparticles. |
| Maleimide-PEG-NHS Heterobifunctional Linker | Creative PEGWorks, Iris Biotech | Enables conjugation of targeting ligands (e.g., peptides, antibodies) to nanoparticle surface for active targeting. |
| Near-IR Fluorophores (DiR, Cy7.5 NHS ester) | Lumiprobe, Thermo Fisher | For in vivo and ex vivo tracking of nanoparticle biodistribution and tumor accumulation. |
| Dialysis Membranes (MWCO 3.5-100 kDa) | Spectrum Labs, Repligen | Purification of nanoparticles from organic solvents and free, unencapsulated API. |
| Dynamic Light Scattering (DLS) / Zeta Potential System | Malvern Panalytical (Zetasizer), Horiba | Core instrument for characterizing nanoparticle size distribution (PDI), zeta potential, and stability. |
| In Vivo Imaging System (IVIS) | PerkinElmer, LI-COR | Non-invasive, longitudinal imaging of fluorescent or luminescent nanoparticle fate in live animal models. |
| Tumor Dissociation Kit (for flow cytometry) | Miltenyi Biotec, STEMCELL Technologies | To dissociate solid tumors into single-cell suspensions for quantifying nanoparticle uptake in specific cell populations. |
Experimental Workflow: From Formulation toIn VivoValidation
A standardized workflow is critical for generating reproducible, publication- and IND-ready data.
Diagram 2: Core R&D Workflow for EPR Nanomedicine
The commercial success of EPR-based polymeric nanomedicines hinges on a dual mastery: advanced nano-formulation science and rigorous navigation of an evolving regulatory framework that demands clear demonstration of therapeutic advantage. Future progress requires a shift from relying on the generalized EPR effect to developing patient-stratified approaches using biomarkers of tumor vasculature, thereby transforming a promising principle into a reliable clinical and commercial reality.
Conclusion
The EPR effect remains a vital, albeit complex, paradigm for designing tumor-targeted polymeric nanoparticles. This synthesis reveals that success hinges on moving beyond a one-size-fits-all reliance on passive accumulation. Future directions must integrate sophisticated, patient-stratified design—combining optimized EPR-compliant physico-chemistry with complementary active targeting and microenvironment-modulating strategies. For researchers, the path forward involves leveraging advanced bio-imaging and predictive models to better understand EPR heterogeneity in patients, while developing next-generation 'smart' polymeric systems with enhanced penetration and controlled release. The ultimate goal is to translate the profound potential of polymeric nanocarriers from a promising research tool into a reliable and impactful clinical reality for oncology and beyond.