Advanced Fabrication Methods for Polymer Nanocomposites: Techniques, Innovations, and Biomedical Applications
This article provides a comprehensive review of contemporary fabrication techniques for polymer nanocomposites, with a specific focus on meeting the needs of researchers and drug development professionals.
Advanced Fabrication Methods for Polymer Nanocomposites: Techniques, Innovations, and Biomedical Applications
Abstract
This article provides a comprehensive review of contemporary fabrication techniques for polymer nanocomposites, with a specific focus on meeting the needs of researchers and drug development professionals. It explores the foundational principles of nanomaterial-polymer interactions, details advanced synthesis methodologies like in situ polymerization and electrospinning, and addresses critical challenges such as nanofiller dispersion and biocompatibility. The content systematically compares the performance outcomes of different fabrication routes and highlights transformative applications in drug delivery, antimicrobial coatings, and tissue engineering. By integrating troubleshooting strategies with a forward-looking perspective, this review serves as a strategic guide for the development of next-generation, high-performance nanocomposites for clinical translation.
The Building Blocks of Polymer Nanocomposites: Nanofillers, Matrices, and Interfacial Science
Definition and Core Significance
Polymer nanocomposites (PNCs) are a class of advanced materials composed of a polymer or copolymer matrix into which nanoscale fillers (typically 1–50 nm in at least one dimension) are dispersed [1] [2]. The fundamental distinguishing factor of PNCs, compared to traditional composites, is the immense amount of interfacial area between the polymer matrix and the nanoscale fillers [1]. This extensive interface is pivotal for transferring the exceptional properties of the nanofillers—such as high strength, electrical conductivity, or thermal stability—to the overall composite, resulting in performance characteristics that often surpass those of conventional materials [1] [3].
The significance of polymer nanocomposites stems from their ability to be uniquely designed for specific applications, offering a combination of properties not found in their individual components [4]. By incorporating small amounts (a few weight percent) of nanofillers, manufacturers can dramatically enhance the polymer's mechanical properties (strength, stiffness, toughness), thermal stability, barrier properties (resistance to gas permeation), and electrical conductivity [1] [4] [3]. This has led to their widespread adoption across a diverse range of industries, from aerospace and automotive to biomedical and electronics.
Types of Nanofillers and Material Properties
Nanofillers are categorized based on their geometry and number of nanoscale dimensions. The selection of nanofiller type is critical for tailoring the properties of the resulting nanocomposite for its intended application [1].
Table 1: Classification and Properties of Common Nanofillers in Polymer Nanocomposites
| Nanofiller Type | Number of Nanoscale Dimensions | Examples | Key Property Enhancements |
|---|---|---|---|
| Nanoplatelets | One (1D) | Nanoclays (e.g., Montmorillonite), Graphene [1] [3] | Improved barrier properties, enhanced flame retardancy, increased stiffness [4] [3] |
| Nanofibers | Two (2D) | Carbon Nanotubes (CNTs), Electrospun Nanofibers [1] [5] | Superior tensile strength, electrical and thermal conductivity, toughness [1] [2] [3] |
| Nanoparticulates | Three (3D) | Nano-Oxides (e.g., SiO₂, TiO₂, ZnO), Metal Nanoparticles (e.g., Silver, Gold) [1] [3] | Antimicrobial properties, UV resistance, catalytic activity, enhanced thermal stability [4] [6] [3] |
Carbon-based nanomaterials, particularly Carbon Nanotubes (CNTs) and graphene, are among the most prominent nanofillers due to their extraordinary combination of mechanical, electrical, and thermal properties [1] [3]. CNTs are further classified into Single-Walled (SWCNTs) and Multi-Walled (MWCNTs), with MWCNTs being more economically feasible for widespread industrial applications [1].
Key Synthesis Techniques and Methodologies
The successful fabrication of high-performance PNCs relies on achieving a homogeneous dispersion of nanofillers within the polymer matrix and ensuring strong interfacial adhesion. Agglomeration of nanoparticles due to strong van der Waals forces is a primary challenge that these synthesis methods aim to overcome [1] [3].
Protocol 3.1: Solution Blending
Principle: This technique involves dispersing the nanofillers and dissolving the polymer in a suitable solvent, followed by mixing and eventual solvent evaporation to form the composite [1].
- Dispersion: The nanofillers are added to a compatible solvent and dispersed using high-shear mixing or ultrasonication to break up agglomerates [1].
- Polymer Introduction: The polymer is dissolved in the same or a compatible solvent to form a separate solution.
- Mixing: The dispersed nanofiller solution and the polymer solution are combined and mixed thoroughly, often with continued mechanical stirring or ultrasonication.
- Solvent Removal: The mixture is cast onto a substrate and the solvent is evaporated, leaving behind a solid nanocomposite film. For complete removal, the film may be placed under vacuum. Considerations: Best for polymers soluble in common solvents. Residual solvent can affect properties, and the process may not be environmentally friendly or economical on a large scale [1].
Protocol 3.2: Melt Blending
Principle: This method involves mixing nanofillers directly with a thermoplastic polymer above its melting temperature using high-shear equipment like a twin-screw extruder [1].
- Drying: The polymer and nanofillers are dried to prevent hydrolysis or degradation during processing.
- Melt Processing: The polymer is fed into an extruder and heated until molten. Nanofillers are fed into the polymer melt via a hopper.
- Shear Mixing: The rotating screws generate high shear forces, which disperse the nanofillers throughout the polymer melt.
- Extrusion and Pelletizing: The homogenized melt is extruded through a die and cooled, then cut into pellets for subsequent shaping (e.g., injection molding). Considerations: Highly compatible with existing industrial infrastructure. Limited to thermoplastics that can withstand the processing temperatures, with potential risk of polymer degradation under high shear [1].
Protocol 3.3: In Situ Polymerization
Principle: Nanofillers are first dispersed in a low-viscosity monomer solution, and the polymerization reaction is initiated to form the polymer matrix around the dispersed fillers [1] [4].
- Monomer Preparation: The monomer and any required catalysts or initiators are prepared.
- Filler Dispersion: The nanofillers are dispersed in the monomer solution using mechanical stirring or ultrasonication.
- Polymerization: Polymerization is initiated thermally, with UV light, or by adding a catalyst, converting the monomer into a solid polymer with embedded nanofillers. Considerations: Excellent for achieving good filler dispersion, especially for insoluble or thermally unstable polymers. Adds complexity due to the chemical process involved [1].
The following workflow diagram illustrates the decision-making process for selecting an appropriate synthesis technique:
The Scientist's Toolkit: Essential Research Reagents and Materials
Fabricating and characterizing polymer nanocomposites requires a range of specialized materials and instruments. The table below details key components essential for research in this field.
Table 2: Essential Research Reagents and Materials for Polymer Nanocomposite Fabrication
| Category | Item / Reagent | Function / Purpose |
|---|---|---|
| Polymer Matrices | Epoxy Resin, Polyamide (Nylon), Polyethylene (PE), Polypropylene (PP), Polybutylene Succinate (PBS) [7] [5] | Serves as the continuous phase or host material; provides bulk form, processability, and determines baseline chemical resistance. |
| Nanofillers | Carbon Nanotubes (SWCNTs/MWCNTs), Nanoclays (Montmorillonite), Graphene, Nano-Oxides (SiO₂, TiO₂, ZnO), Metal Nanoparticles (Ag) [3] [5] | The reinforcing phase; imparts enhanced mechanical, thermal, electrical, or barrier properties to the composite. |
| Solvents | Tetrahydrofuran (THF), Dimethylformamide (DMF), Chloroform, Toluene | Dissolves polymer for solution processing; medium for dispersing nanofillers. |
| Functionalization Agents | Silane coupling agents, Surfactants | Chemically modifies nanofiller surface to improve compatibility and dispersion within the polymer matrix [1]. |
| Processing Aids | Plasticizers, Thermal Stabilizers | Improves processability and prevents thermal degradation during high-temperature processing like melt blending. |
| Characterization Equipment | Scanning Electron Microscope (SEM), Transmission Electron Microscope (TEM), X-ray Diffractometer (XRD), Spectroscopic Ellipsometry | Analyzes morphology, dispersion state, crystalline structure, and thermal transitions of the nanocomposite [7]. |
Applications and Market Impact
The global market for polymer nanocomposites was valued at $14.61 billion in 2024 and is projected to grow rapidly to $32.39 billion by 2029, reflecting a compound annual growth rate (CAGR) of 17.8% [5]. This growth is driven by increasing demand across multiple high-tech industries.
Table 3: Key Application Areas for Polymer Nanocomposites
| Industry Sector | Specific Applications | Key Property Utilized |
|---|---|---|
| Automotive & Aerospace | Interior/exterior parts, engines/powertrains, suspensions, lightweight structural components [5] | Reduced weight, improved mechanical strength, enhanced thermal stability [8] [5] |
| Electronics & Electricals | Conductive films, sensors, thermal management systems, organic solar cells [4] [6] | Enhanced electrical conductivity, thermal stability, unique optical properties [6] [3] |
| Biomedical & Healthcare | Drug delivery systems, tissue engineering scaffolds, biosensors, biomedical implants [4] [2] [3] | Biocompatibility, tailored functionality, high drug-loading capacity, improved cell attachment [4] [2] |
| Packaging | Food packaging, barrier films | Improved gas/water vapor barrier properties, antimicrobial activity, mechanical strength [4] [5] |
| Energy | Solar cells, energy storage devices | Enhanced light absorption, charge transport, and storage capabilities [6] |
In the biomedical field, PNCs are particularly transformative. They act as sophisticated nanocarriers for targeted drug delivery, protecting therapeutic agents and controlling their release kinetics at specific sites in the body, thereby minimizing side effects [4] [2]. In tissue engineering, nanofibrous PNC scaffolds provide a favorable environment for cell attachment and growth, facilitating the regeneration of tissues such as bone and nerve [4] [2]. The following diagram outlines the primary biomedical applications of polymer nanocomposites:
Polymer nanocomposites (PNCs) represent a advanced class of materials where nanofillers, with at least one dimension in the 1-100 nanometer range, are incorporated into a polymer matrix to dramatically enhance its intrinsic properties [1] [9]. The foundation of this enhancement lies in the nanoscale interactions between the polymer and the filler. Unlike traditional microcomposites, which require high filler loading (20-30% by weight) for modest improvement, nanocomposites achieve superior reinforcement at significantly lower loadings (3-5% by weight) due to the exceptionally high specific surface area of the nanofillers, which can be as large as 2630 m²/g for materials like graphene [10] [11]. This high surface area enables profound interfacial interactions, leading to unprecedented improvements in mechanical strength, thermal stability, electrical conductivity, and barrier properties without compromising the polymer's processability or increasing its density [12] [9].
The dispersion state of the nanofiller is a critical determinant of the nanocomposite's final performance. Ideally, nanoparticles should be uniformly dispersed and individually coated by the polymer to achieve optimal load transfer and uniform stress distribution [11]. Three primary morphologies are possible: conventional composites (phase-separated, with properties similar to traditional composites), intercalated nanocomposites (where polymer chains insert between filler layers, creating a well-ordered multilayer structure), and exfoliated nanocomposites (where filler layers are completely and uniformly separated and dispersed in the polymer matrix) [9]. The exfoliated structure provides maximum reinforcement due to the largest possible interfacial area between the matrix and the nanoparticles [9].
Classification and Properties of Key Nanofillers
Nanofillers can be systematically classified into three broad categories based on their composition and origin: carbon-based, metal/oxide, and organic nanomaterials. The table below provides a comparative overview of the primary nanofiller types, their key properties, and common applications.
Table 1: Classification and Characteristics of Key Nanofillers
| Nanofiller Category | Specific Types | Key Properties | Common Applications |
|---|---|---|---|
| Carbon-Based | Carbon Nanotubes (CNTs) [12] [13] | Exceptional mechanical strength (Tensile strength: 50-150 GPa, Modulus: ~1 TPa), high electrical & thermal conductivity [13] [11] | Aerospace, automotive, sensors, supercapacitors, electromagnetic absorbers [13] [14] |
| Graphene [10] [11] | High surface area (2630 m²/g), excellent electrical conductivity (6000 S/cm), superior thermal conductivity (~5000 W/m·K), gas impermeability [10] [11] | Advanced composites, energy storage, sensors, gas barrier coatings [10] [9] | |
| Fullerenes [12] | Unique cage-like structure, good electron acceptor properties | Pharmaceutical, cosmetic additives, organic photovoltaics | |
| Metal/Oxide | Metal Oxides (e.g., ZnO, TiO₂, Al₂O₃, CuO) [15] [16] | Good thermal stability, photocatalytic activity (TiO₂), UV-blocking, antimicrobial properties (ZnO, CuO) [15] | Food packaging, antimicrobial coatings, biomedical implants, catalysts, solar cells [15] [16] |
| Nanoclays (e.g., Montmorillonite) [9] [11] | Layered silicate structure, high modulus (178-265 GPa), excellent barrier properties, flame retardancy [11] | Automotive parts, food packaging, flame-retardant materials [9] [11] | |
| Metal Nanoparticles (e.g., Ag, Au, Fe₃O₄) [16] | Unique optical properties (Surface Plasmon Resonance), magnetic properties (Fe₃O₄), antimicrobial activity (Ag) | Drug delivery, biosensors, medical imaging, catalysis [16] | |
| Organic | Nanocellulose (CNC, CNF) [10] [14] | Biodegradability, biocompatibility, low density, high strength, good stiffness [10] [14] | Biomedical applications, biodegradable packaging, reinforcement in bioplastics [15] [14] |
| Dendrimers [10] [16] | Highly branched, tree-like structure, monodisperse size, tunable surface functionality | Drug delivery, gene therapy, imaging agents [10] [16] | |
| Liposomes/Micelles [16] | Hollow sphere structure, ability to encapsulate hydrophilic/hydrophobic substances | Targeted drug delivery, nanoreactors [16] |
The following diagram illustrates the hierarchical classification of these nanofillers and their structural relationships.
Experimental Protocols for Nanocomposite Fabrication and Characterization
Fabrication Methods
The successful integration of nanofillers into a polymer matrix is crucial for achieving the desired properties in the nanocomposite. The following protocols detail the most common and effective fabrication methods.
Protocol 1: Solution Blending Method
Principle: This method involves dispersing the nanofiller in a suitable solvent, mixing it with a polymer solution, and subsequently removing the solvent to form the composite [1] [17].
Step-by-Step Procedure:
- Polymer Dissolution: Dissolve the selected polymer (e.g., Polyvinyl alcohol, Polystyrene) in an appropriate solvent (e.g., water, toluene, chloroform) under constant magnetic stirring at 40-60°C until a homogeneous solution is obtained.
- Nanofiller Dispersion: In a separate container, disperse the predetermined weight percentage (e.g., 0.5-5 wt%) of nanofiller (e.g., CNTs, graphene oxide) in the same solvent. Subject this mixture to probe ultrasonication (e.g., 300-500 W) for 15-60 minutes in an ice bath to prevent solvent evaporation and agglomerate breakdown.
- Mixing: Combine the polymer solution and the nanofiller dispersion. Stir the mixture vigorously for 1-2 hours, followed by additional bath ultrasonication for 30 minutes to ensure homogeneous distribution.
- Solvent Removal: Cast the final mixture into a petri dish and allow the solvent to evaporate at room temperature or in a vacuum oven at elevated temperature (e.g., 50°C) for 12-24 hours to ensure complete solvent removal.
- Post-Processing: The resulting solid nanocomposite film can be further dried and cut for testing.
Advantages and Limitations:
- Advantages: Simple, effective for lab-scale production, allows for good control over filler dispersion, suitable for polymers with low thermal stability [1].
- Limitations: Use of large quantities of solvent (economic and environmental concerns), potential for residual solvent affecting properties, not easily scalable for industrial production [1].
Protocol 2: Melt Blending Method
Principle: This method involves the direct mechanical mixing of nanofillers with a molten thermoplastic polymer under high shear forces, typically using an internal mixer or a twin-screw extruder [1] [17].
Step-by-Step Procedure:
- Drying: Pre-dry both the polymer pellets (e.g., Polypropylene, Nylon) and the nanofiller in a vacuum oven at 80°C for at least 6 hours to remove moisture.
- Melt Processing: Feed the pre-mixed polymer and nanofiller into a twin-screw extruder or an internal mixer (e.g., Haake Rheomix). The processing temperature should be set above the glass transition temperature (Tg) for amorphous polymers or the melting temperature (Tm) for semicrystalline polymers.
- Shear Mixing: Process the mixture at a specified rotor speed (e.g., 50-100 rpm) and temperature profile for a residence time of 5-10 minutes to ensure sufficient shear stress for de-agglomeration and dispersion of the nanofiller.
- Pelletization and Molding: The extruded strand is water-cooled and pelletized. The pellets are then injection-molded or compression-molded into standard test specimens (e.g., ASTM dog-bone shapes) for characterization.
Advantages and Limitations:
- Advantages: Solvent-free, environmentally friendly, compatible with existing industrial equipment (high scalability), simplicity [1] [17].
- Limitations: Limited to processable thermoplastics, potential for polymer degradation at high shear and temperature, may be less effective for achieving exfoliation compared to solution methods [1].
Protocol 3: In Situ Polymerization Method
Principle: In this method, nanofillers are first dispersed in a low-viscosity monomer or monomer solution, followed by the polymerization of the monomer around the filler [1] [17].
Step-by-Step Procedure:
- Monomer-Filler Dispersion: Add the nanofiller to the liquid monomer (e.g., ε-caprolactam for Nylon 6, methyl methacrylate for PMMA). Disperse the filler using magnetic stirring and ultrasonication for 30-60 minutes.
- Polymerization Initiation: Transfer the mixture to a reaction vessel. Add the required initiator or catalyst under an inert atmosphere (e.g., Nitrogen or Argon gas).
- Polymerization: Carry out the polymerization reaction at a specified temperature and time according to the polymer system (e.g., 100°C for 12-24 hours).
- Product Recovery: After polymerization, the resulting nanocomposite solid is crushed, washed, and dried to remove any unreacted monomer or by-products.
Advantages and Limitations:
- Advantages: Enables excellent filler dispersion and strong filler-matrix interaction, suitable for insoluble or thermally unstable polymers, monomers can infiltrate filler agglomerates [1] [17].
- Limitations: Complexity of the chemical process, requires precise control over polymerization parameters, may not be universally applicable to all polymer systems [1].
Characterization Techniques
Confirming the structure, morphology, and properties of the synthesized nanocomposites is a critical step. The following workflow outlines the standard characterization pathway.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists essential materials, reagents, and equipment required for the fabrication and characterization of polymer nanocomposites, as derived from the experimental protocols.
Table 2: Essential Research Reagents and Equipment for Nanocomposite Fabrication
| Category | Item | Specification / Examples | Primary Function |
|---|---|---|---|
| Polymer Matrices | Thermoplastics | Polypropylene (PP), Nylon 6 (PA6), Polyethylene terephthalate (PET), Thermoplastic Polyurethane (TPU) [14] [9] | Primary matrix; determines bulk processability and properties. |
| Thermosets | Epoxy, Phenol-formaldehyde (PF) [13] [9] | Cross-linked matrix for high-performance applications. | |
| Biopolymers | Chitosan, Poly(lactic acid) (PLA), Starch, Poly(vinyl alcohol) (PVA) [15] [9] | Sustainable and biocompatible matrix for green composites. | |
| Nanofillers | Carbon-Based | SWCNTs, MWCNTs, Graphene nanoplatelets, Graphene Oxide [12] [13] [11] | Provide electrical/thermal conductivity and mechanical reinforcement. |
| Metal/Oxide | ZnO nanoparticles, TiO₂ nanoparticles, CuO nanoparticles, Montmorillonite clay [15] [11] | Impart UV resistance, photocatalytic activity, antimicrobial properties, and barrier improvement. | |
| Organic | Nanocrystalline Cellulose (NCC), Dendrimers [10] [14] | Provide reinforcement in bio-nanocomposites or act as carriers for drug delivery. | |
| Solvents & Chemicals | Dispersion Solvents | Deionized Water, Toluene, Chloroform, Dimethylformamide (DMF) [1] [11] | Medium for solution blending and filler dispersion. |
| Surfactants | Sodium Dodecyl Sulfate (SDS), Sodium Dodecyl Benzenesulfonate (SDBS) [11] | Stabilize nanoparticle dispersion and prevent re-agglomeration. | |
| Coupling Agents | (3-Aminopropyl)triethoxysilane (APTES), other silanes [17] | Chemically modify filler surface to improve compatibility with polymer matrix. | |
| Equipment | Dispersion | Ultrasonic Bath, Ultrasonic Probe Homogenizer [1] [11] | Apply mechanical energy to break up nanofiller agglomerates. |
| Mixing & Processing | Twin-Screw Extruder, Internal Mixer (e.g., Haake Rheomix), Magnetic Stirrer/Hotplate [1] [17] | Melt and homogenize the polymer-nanofiller mixture. | |
| Molding | Injection Molding Machine, Compression Press | Shape the final nanocomposite into test specimens or products. | |
| Characterization | Tensile Testing Machine, TGA, DSC, SEM, TEM, XRD, FTIR [9] | Analyze morphology, structure, and properties of the final nanocomposite. |
The strategic classification of nanofillers into carbon-based, metal/oxide, and organic categories provides a structured framework for researchers to select the most appropriate material for tailoring the properties of polymer nanocomposites. The efficacy of the final material is contingent upon a deep understanding of the intrinsic properties of the nanofiller and the selection of a suitable fabrication protocol—be it solution blending, melt blending, or in situ polymerization—to overcome the perennial challenge of achieving homogeneous dispersion and strong interfacial adhesion. As this field advances, the focus is shifting towards sustainable and functional nanocomposites, particularly those utilizing biopolymers and organic nanofillers like nanocellulose for biomedical and green packaging applications [15] [10]. Furthermore, the development of hybrid nanocomposites, which combine multiple nanofillers such as polymer/fiber/CNT systems, presents a promising frontier for creating multifunctional materials with synergistic properties that meet the demanding requirements of next-generation applications in aerospace, automotive, and advanced biomedicine [13]. The ongoing research into more efficient, scalable, and eco-friendly fabrication methods will be pivotal in unlocking the full commercial potential of these advanced materials.
The selection of an appropriate polymer matrix is a foundational step in the design of polymer nanocomposites (PNCs) for advanced applications, including drug delivery and biomedical devices. The polymer matrix serves as the continuous phase that hosts the nanoscale fillers, critically determining the composite's mechanical integrity, degradation profile, biocompatibility, and overall functionality [4]. The core dichotomy in selection lies between natural polymers, prized for their innate bioactivity and compatibility, and synthetic polymers, which offer superior and tunable mechanical properties and processability [18]. A third, rapidly evolving category is that of biodegradable polymers, both natural and synthetic, which are engineered to break down into environmentally benign or metabolizable byproducts after their useful life [19] [20]. This document provides structured application notes and experimental protocols to guide researchers in selecting and working with these polymer matrices within the context of nanocomposites fabrication.
Comparative Analysis of Polymer Matrices
A systematic selection of a polymer matrix requires a clear understanding of the properties and trade-offs associated with each class. The following tables summarize the key characteristics, advantages, and limitations of natural, synthetic, and biodegradable polymers.
Table 1: Comparison of Natural and Synthetic Polymer Matrices
| Aspect | Natural Polymers | Synthetic Polymers |
|---|---|---|
| Origin | Extracted from biomass (e.g., plants, animals, microorganisms) [20]. | Chemically synthesized from petroleum-based or renewable monomers [20]. |
| Key Examples | Collagen, chitosan, alginate, starch, cellulose, silk fibroin [18] [20]. | Poly(lactic acid) (PLA), Poly(glycolic acid) (PGA), Poly(ε-caprolactone) (PCL), Poly(ethylene glycol) (PEG) [18] [20]. |
| Biocompatibility | Typically excellent, due to biological recognition [18]. | Variable; can be designed to be high, but may elicit inflammatory responses [18]. |
| Biodegradability | Inherently biodegradable; enzymatic degradation [20]. | Not inherent; must be engineered (e.g., aliphatic polyesters) [19] [20]. |
| Mechanical Properties | Often limited; can be weak and brittle [18]. | Broadly tunable; can achieve high strength, toughness, and elasticity [18]. |
| Processability & Reproducibility | Can be challenging; batch-to-batch variability is common [18]. | Excellent; highly reproducible with consistent properties [18]. |
| Cost | Often lower raw material cost, but purification can be expensive [21]. | Can be cost-effective at scale, though some high-performance polymers are expensive [21]. |
Table 2: Properties of Common Biodegradable Polymers for Nanocomposites
| Polymer | Origin | Tensile Strength (MPa) | Young's Modulus (GPa) | Melting Temp. (°C) | Degradation Time | Key Applications |
|---|---|---|---|---|---|---|
| PGA [20] | Synthetic | 70 - 117 | 6.1 - 7.2 | 220 - 231 | Months | Sutures, tissue scaffolds |
| PLA [20] | Synthetic | 48 - 53 | 3.1 - 3.6 | 170 - 180 | 12-24 months | Packaging, medical devices |
| PCL [18] | Synthetic | 20 - 42 | 0.3 - 0.5 | 58 - 65 | 2-3 years | Long-term implants, drug delivery |
| Thermoplastic Starch [20] | Natural | 16 - 22 | N/A | N/A | Variable | Bio-based packaging, agriculture |
| Chitosan [18] | Natural | N/A | N/A | N/A | Enzyme-dependent | Wound dressings, drug carriers |
Experimental Protocols for Nanocomposite Fabrication and Testing
Protocol: Fabrication of a Biodegradable Polymer/Nanoclay Nanocomposite via Melt Blending
Principle: This protocol describes the integration of nanoclays into a thermoplastic biodegradable polymer (e.g., PLA) using melt blending, a common and scalable industrial method. The high shear forces at elevated temperatures disperse the nanofillers within the polymer matrix [22].
Materials:
- Polymer Matrix: Polylactic acid (PLA) pellets.
- Nanofiller: Organically modified Montmorillonite (MMT) nanoclay.
- Equipment: Twin-screw extruder, injection molding machine, vacuum oven.
Procedure:
- Drying: Dry PLA pellets and nanoclay in a vacuum oven at 80°C for at least 12 hours to remove moisture.
- Pre-mixing: Manually pre-mix the dried PLA pellets with nanoclay (e.g., at 3-5 wt%) in a container to ensure initial homogeneity.
- Melt Compounding: Feed the pre-mixed material into a co-rotating twin-screw extruder. Typical processing parameters for PLA:
- Temperature Profile: 170°C (hopper) to 190°C (die).
- Screw Speed: 100-200 rpm.
- Maintain a consistent feed rate to ensure proper shear and residence time.
- Pelletizing: The extruded strand is water-cooled and pelletized to form the masterbatch of nanocomposite.
- Specimen Fabrication: Process the pellets using an injection molding machine to form standard test specimens (e.g., ASTM D638 tensile bars). The molding temperature should be slightly below the extrusion temperature to prevent degradation.
Protocol: Assessment of Polymer Nanocomposite Biodegradation
Principle: This protocol outlines a method to evaluate the biodegradation rate of a nanocomposite under controlled composting conditions, simulating an industrial composting environment [19] [20].
Materials:
- Test Material: Nanocomposite specimens (e.g., 20 mm x 20 mm x 1 mm sheets).
- Control: Cellulose (positive control), polyethylene (negative control).
- Inoculum: Mature compost from municipal solid waste, sieved to ≤ 10 mm.
- Equipment: Bioreactors or glass jars, controlled temperature chamber, analytical balance, pH meter.
Procedure:
- Preparation: Weigh the initial mass (W₀) of each test specimen. Characterize the compost inoculum for pH and dry solid content.
- Setup: Mix the solid inoculum with the test and control materials in bioreactors. The test material concentration should be less than 1% on a dry solids basis. Maintain the compost moisture content at around 50-55%.
- Incubation: Incubate the bioreactors in the dark at a constant thermophilic temperature of 58°C ± 2°C. This is a standard condition for industrial composting [19].
- Aeration & Monitoring: Aerate the compost periodically with CO₂-free air to maintain aerobic conditions. Monitor the temperature, and adjust the moisture content with deionized water as needed.
- Analysis:
- Biodegradation Rate: Measure the evolved CO₂ periodically using titration or GC analysis. The percentage of biodegradation is calculated as the ratio of the measured CO₂ from the test material to the theoretical maximum CO₂ it could produce.
- Material Analysis: At the end of the test (typically 180 days), retrieve the specimens. Carefully clean and dry them to determine the residual dry mass (Wᵣ). Calculate the percentage of mass loss: [(W₀ - Wᵣ) / W₀] x 100. Analyze the surface morphology of retrieved specimens using Scanning Electron Microscopy (SEM) to observe physical deterioration and microbial colonization.
Decision Workflow and Experimental Process Visualization
The following diagrams, generated using DOT language, illustrate the logical decision-making process for polymer selection and the experimental workflow for nanocomposite fabrication and testing.
Polymer Matrix Selection Workflow
Nanocomposite Fabrication and Testing Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Polymer Nanocomposites Research
| Reagent/Material | Function & Application | Key Considerations |
|---|---|---|
| Polylactic Acid (PLA) [20] [21] | A versatile, bio-based, biodegradable synthetic polymer matrix. Used in packaging, biomedical scaffolds, and 3D printing. | Exists in L- and D- isomers; ratio affects crystallinity and degradation rate. Can be brittle; often blended or plasticized. |
| Chitosan [18] | A natural cationic polymer derived from chitin. Excellent for wound healing dressings and drug delivery due to its hemostatic and antimicrobial properties. | Solubility is highly dependent on pH and degree of deacetylation. Viscosity is molecular weight dependent. |
| Polyhydroxyalkanoates (PHA) [19] [21] | A family of natural, biodegradable polyesters produced by microorganisms. Used in biocomposites for medical implants and packaging. | Properties vary widely with monomer composition. Can be expensive to produce at scale. |
| Joncyl ADR Series [21] | A commercial styrene-acrylic multi-functional epoxide oligomer used as a reactive compatibilizer and chain extender in PLA/PBAT and other blends. | Improves miscibility and interfacial adhesion in polymer blends, leading to enhanced mechanical properties and melt strength. |
| Maleic Anhydride [21] | A chemical used to graft onto polymer chains (e.g., PLA, PP) to create compatibilizers for natural fibre-reinforced composites. | The grafted maleic anhydride groups can react with hydroxyl groups on natural fibres, improving fibre-matrix bonding. |
| Montmorillonite Clay [4] [3] | A layered silicate nanoclay used as a nanofiller to improve mechanical strength, thermal stability, and barrier properties of polymer matrices. | Must be organically modified to become organophilic and achieve proper exfoliation/dispersion in most polymer matrices. |
| Graphene Oxide (GO) [4] [3] | A carbon-based nanomaterial used to enhance electrical conductivity, mechanical strength, and thermal stability of composites. | High surface area and functional groups facilitate dispersion and interaction with the polymer matrix. Can be reduced to conductive graphene. |
In polymer nanocomposites (PNCs), the interface between the nanoscale filler and the polymer matrix is not merely a boundary but a dynamic, three-dimensional region that dictates ultimate material performance [23]. The fundamental challenge in PNC design lies in overcoming inherent thermodynamic barriers to achieve uniform nanoparticle dispersion and strong interfacial adhesion [24]. When the interface is optimally engineered, it facilitates efficient stress transfer, modifies local polymer chain dynamics, and enhances barrier properties, leading to unprecedented improvements in mechanical, thermal, and functional characteristics [23]. This document provides detailed application notes and experimental protocols for characterizing and manipulating interfacial interactions in polymer nanocomposites, with specific methodologies relevant to advanced materials research and development.
Key Concepts & Quantitative Data
The Critical Role of the Interfacial Region
The interfacial region in PNCs consists of polymer chains whose conformation, mobility, and density are altered by interactions with the nanoparticle surface [23]. Traditionally, polymers adsorbed onto nanoparticles exhibit slowed relaxation and form a high-density, immobilized "dead layer" that can embrittle the composite [23]. Recent research demonstrates that by engineering the architecture of the interfacial polymers—specifically by creating bound polymer loops (BLs) on nanoparticle surfaces—it is possible to design relaxation-enhanced PNCs. These materials exhibit simultaneously improved processability (reduced melt viscosity) and enhanced mechanical properties (increased strength and toughness) in the glassy state [23].
Quantifying Interfacial Effects on Composite Properties
Table 1: Experimental Data on Interfacial Engineering in Polymer Nanocomposites
| Material System | Interfacial Engineering Strategy | Key Quantitative Results | Citation |
|---|---|---|---|
| Polymer-Polymer Composite | Cold drawing of polymer blend to create nanofibrils; "creating instead of adding" | Tensile strength and modulus increases of 300-400%; up to tenfold improvement over older techniques [24] | [24] |
| Polystyrene/Silica PNC | Introduction of bound polystyrene loops (6 nm thick) on silica nanoparticle surfaces | Formation of a dynamic, loose particle network; maintained fluid-like, low-viscosity dynamics at high NP loading; glassy materials showed enhanced toughness and strength [23] | [23] |
| Epoxy/Magnetic NP PNC | Bulk & surface modification: Co-doping + PEG-functionalization of Fe₃O₄ NPs | Achieved "Excellent" cure state (Cure Index) at 0.1 wt.% loading; higher activation energy vs. neat epoxy [25] | [25] |
| PLA/PCL/Silica Blend | Use of nano-silica (Aerosil200) as compatibilizer | SEM-EDS showed Si concentration up to 10x nominal value at protrusions; encapsulation by PCL; increased contact angle (more hydrophobic) [26] | [26] |
| Concrete Nanocomposite Coating | Organoclay (Cloisite 30B) in acrylic-fluorinated resin at 2-6 wt.% | Enhanced barrier properties: reduced water transport, improved sulfate attack resistance; low impact on color [27] | [27] |
Experimental Protocols
Protocol: Fabrication of "Genuine" Nanofibrillar Polymer-Polymer Composites
This protocol outlines a method to bypass dispersion challenges by creating nanofibrils in situ during processing [24].
3.1.1. Research Reagent Solutions
Table 2: Essential Materials for Nanofibrillar Composite Fabrication
| Item | Function/Description |
|---|---|
| Polymer Blend Components | A two-polymer system where the minor component can be transformed into a fibrillar morphology. |
| Melt Blending Equipment | (e.g., Twin-screw extruder) For initial homogenization of the polymer blend. |
| Cold Drawing Apparatus | Equipment to uniaxially stretch the solid blend below the melting point of the minor component. |
| Selective Solvent | A chemical that selectively dissolves the matrix polymer to isolate nanofibrils for characterization. |
3.1.2. Methodology
- Melt Blending: Process the selected polymer components under controlled temperature and shear rate conditions to form an initial blend with a dispersed phase of the minor component [24].
- Cold Drawing: Subject the solid blend to uniaxial stretching at a temperature below the melting point of the minor component but above the glass transition temperature of both polymers. This process deforms the dispersed phase into uniformly dispersed nanofibrils [24].
- Formation of Composite:
- Path A: Nanofibrillar Polymer-Polymer Composite: The cold-drawn material, containing nanofibrils of one polymer within the matrix of the other, is the final composite product [24].
- Path B: Nanofibrillar Single Polymer Composite: The matrix polymer is selectively extracted from the cold-drawn blend, leaving behind neat nanofibrils. These nanofibrils are then processed (e.g., via compression molding) to form a composite where the fibrils reinforce a matrix of the same polymer [24].
3.1.3. Workflow Visualization
Protocol: Designing Relaxation-Enhanced PNCs with Bound Polymer Loops
This protocol details the molecular design of nanoparticle interfaces to enhance polymer dynamics and composite performance [23].
3.2.1. Research Reagent Solutions
Table 3: Essential Materials for Relaxation-Enhanced PNCs
| Item | Function/Description |
|---|---|
| Silica Nanoparticles (SiOx) | Model filler (e.g., 65 ± 10 nm diameter). |
| Statistical Copolymer | Poly(styrene-ran-4-hydroxystyrene) [P(S-ran-HS)] of varying HS mole fractions (f_HS). |
| Matrix Polymer | e.g., Polystyrene (PS), M_w = 370 kg mol⁻¹. |
| Solvents | Methyl ethyl ketone (MEK), Toluene, Chloroform. |
| Annealing Oven | For thermal treatment under vacuum. |
3.2.2. Methodology
Preparation of Loop-Covered Nanoparticles:
- Disperse silica nanoparticles in a solution of P(S-ran-HS) in methyl ethyl ketone.
- Cast and dry the composite dispersion.
- Anneal the resulting solid composite at
T_g + 50 °C(e.g., 150 °C for PS) for 24 hours under vacuum. This promotes the adsorption of the 4-hydroxystyrene (HS) segments onto the nanoparticle surface via H-bonding with surface silanol groups, creating bound loops (BLs) of the polystyrene segments [23]. - Remove non-attached polymer chains by solvent leaching with chloroform to obtain the final BL–SiOx nanoparticles [23].
Characterization of Bound Loops:
- Use Transmission Electron Microscopy (TEM) to visualize and measure the thickness of the bound loop layer (hBL). The thickness decreases with increasing HS mole fraction (fHS) [23].
- Perform Thermogravimetric Analysis (TGA) to quantify the grafting density of the polymer loops.
- Employ Solid-state Proton NMR (¹H-NMR) on the final composite melt to confirm enhanced molecular mobility of the bound loops compared to densely adsorbed layers [23].
Fabrication and Testing of PNCs:
- Incorporate the BL–SiOx nanoparticles into the PS matrix via solution casting.
- Perform rheological measurements on the molten PNCs to demonstrate reduced viscosity and enhanced flowability.
- Conduct mechanical tests on the glassy PNCs to confirm enhanced toughness and strength [23].
3.2.3. Workflow Visualization
Characterization Techniques for Interfacial Analysis
A multi-technique approach is essential to fully characterize the interface in PNCs.
- Scanning Electron Microscopy with Energy-Dispersive X-ray Spectroscopy (SEM-EDS): Provides quantitative analysis of filler distribution and location within a polymer blend. EDS mapping can reveal component affinity, such as the encapsulation of nano-silica by PCL in a PLA matrix [26].
- Solid-state Proton NMR (¹H-NMR): Directly probes the molecular mobility of polymers at the interface in the composite melt, differentiating the dynamics of bound loops from immobilized adsorbed layers [23].
- X-ray Reflectivity (XRR): Measures the density profile of interfacial polymer layers, confirming the presence or absence of a high-density "dead layer" [23].
- Rheology: The temperature-dependent shifting factors (a_T) from rheological measurements are sensitive to the constrained dynamics of interfacial polymers and are a key indicator of the success of relaxation-enhancement strategies [23].
- Water Contact Angle Measurements: Used to determine changes in surface free energy and hydrophobicity/hydrophilicity induced by the addition of nanofillers (e.g., increased hydrophobicity with nano-silica) [26].
The performance of polymer nanocomposites is profoundly governed by the properties of the polymer-filler interface. Strategic interfacial engineering moves beyond simple mixing to create advanced materials with tailored properties. The protocols outlined herein—ranging from the fabrication of genuine nanocomposites via cold drawing to the molecular design of relaxation-enhanced interfaces with bound polymer loops—provide a robust toolkit for researchers. The critical insights are that optimizing the nanostructure of the interface can break traditional trade-offs, leading to materials that are both easier to process and mechanically superior [24] [23]. Success in this field relies on the rigorous application of the quantitative characterization techniques described to validate interfacial design and its direct link to macroscopic performance.
Polymer nanocomposites represent a advanced class of materials formed by dispersing nanoscale fillers into polymer matrices, resulting in synergistic property enhancements far exceeding conventional composite performance. These materials leverage the unique effects emerging at the nanoscale to overcome inherent limitations of polymers while introducing new functionalities. Within the broader thesis on polymer nanocomposites fabrication methods, this application note systematically examines the fundamental mechanisms governing the enhancement of electrical, mechanical, and thermal properties. We provide researchers with structured quantitative data, detailed experimental protocols, and visual workflows to facilitate the rational design and fabrication of advanced polymer nanocomposites for specialized applications across electronics, aerospace, and biomedical fields.
Electrical Property Enhancement
Percolation and Conductive Network Formation
The electrical property enhancement in conductive polymer nanocomposites primarily follows a percolation phenomenon, where a continuous conductive network forms throughout the insulating polymer matrix at a critical filler concentration known as the percolation threshold [28]. Below this threshold, the composite remains insulating; above it, electrical conductivity increases dramatically by several orders of magnitude.
Carbon-based nanofillers, particularly carbon black (CB), carbon nanotubes (CNTs), and graphene nanoplatelets (GnP), excel as conductive fillers due to their high intrinsic conductivity, stability against oxidation, and ability to form interconnected networks at low loadings [28] [29]. The nanoscale dimensions and high aspect ratio of these fillers enable electron transport through tunneling effects between adjacent particles, significantly reducing the percolation threshold compared to conventional micron-sized fillers.
Table 1: Electrical Property Enhancement in Polymer Nanocomposites
| Nanocomposite System | Filler Content | Electrical Conductivity | Enhancement Factor | Key Mechanism |
|---|---|---|---|---|
| PLA/CNT [30] | 3 wt% | - | 11,400% (at 100 kHz) | Formation of conductive percolating network |
| GnP/LCP [29] | 5 wt% | 0.05 S/m | - | Homogeneous dispersion & platelet alignment |
| CB/Polysiloxane [28] | 1 wt% | - | - | Pulsed electric field-induced chain formation |
External Field-Assisted Alignment
A significant advancement in controlling percolation structures involves applying external fields during processing. Traditional DC electric fields face limitations due to dielectric breakdown at high voltages [28]. The innovative use of nanosecond pulsed electric fields allows the application of substantially higher field strengths (up to 7500 V/mm in one study) without causing material breakdown, enabling precise control over the formation of linear CB percolation networks [28].
Diagram 1: This workflow illustrates the formation of conductive percolation networks in a polymer nanocomposite under a nanosecond pulsed electric field.
Experimental Protocol: Nanosecond Pulsed Electric Field Alignment
Objective: To induce controlled percolation structures of carbon black in a polysiloxane matrix using a nanosecond pulsed electric field.
Materials:
- Conductive Filler: Carbon Black (CB), average diameter 50 nm, resistivity 100 Ω·m
- Polymer Matrix: Polysiloxane prepolymer (e.g., Part A: low viscosity ~0.1-0.2 Pa·s; Part B: high viscosity ~0.8-1.0 Pa·s)
- Solvent: Toluene for initial dispersion
Equipment:
- Nanosecond pulsed power generator (e.g., semiconductor switching devices like MOSFETs)
- Custom parallel-plate electrode cell (copper electrodes)
- Vacuum oven for solvent removal and curing
- Scanning Electron Microscope (SEM) for characterization
- Impedance analyzer for electrical conductivity measurement
Procedure:
- Solution Preparation: Dissolve the polysiloxane prepolymer Part A in toluene using a magnetic stirrer. Add 1 wt% CB relative to the total polymer weight and disperse using high-shear mixing (e.g., 2000 rpm for 10 minutes) followed by bath sonication for 30 minutes.
- Solvent Evaporation: Remove toluene by heating the mixture at 60°C with continuous stirring until a viscous, solvent-free suspension is obtained.
- Crosslinker Addition: Mix the CB/Part A suspension with polysiloxane Part B (crosslinker) in a 1:1 weight ratio. Degas the mixture under vacuum to remove air bubbles.
- Electric Field Application: Pour the mixture into the parallel-plate electrode cell. Apply the nanosecond pulsed electric field with the following parameters:
- Field strength: 1875 - 7500 V/mm
- Pulse width: 200 ns
- Pulse repetition frequency: 100 Hz
- Application time: Maintain field during crosslinking (typically 1-2 hours at room temperature)
- Curing: Allow the composite to fully crosslink under the applied field.
- Characterization: Analyze the resulting percolation structure using SEM and measure electrical conductivity via a four-point probe method or impedance analyzer.
Key Parameters:
- The viscosity of the polymer matrix must be optimized to allow filler alignment while preventing sedimentation.
- Field strength must remain below the dielectric breakdown threshold of the composite.
Mechanical Property Enhancement
Reinforcement Mechanisms
The incorporation of nanoscale fillers such as graphene, CNTs, and ceramic nanoparticles enhances mechanical properties through several mechanisms:
- Stress Transfer Efficiency: The high surface-area-to-volume ratio of nanofillers creates an extensive polymer-filler interface, enabling efficient stress transfer from the polymer matrix to the stiffer reinforcement phase [31]. For instance, functionalized graphene/polymer nanocomposites have demonstrated 57% and 70% enhancements in Young's modulus and tensile strength, respectively [31].
- Crack Deflection and Barrier Effects: Well-dispersed nanoparticles act as physical obstacles to propagating cracks, forcing them to deviate from their path and thereby increasing the fracture energy [32].
- Constrained Polymer Dynamics: Nanoparticles restrict the mobility of surrounding polymer chains, particularly in the interfacial region, leading to increased modulus [32].
Table 2: Mechanical Property Enhancement in Polymer Nanocomposites
| Nanocomposite System | Filler Content | Tensile Strength | Young's Modulus | Key Mechanism |
|---|---|---|---|---|
| PLA/CNT [30] | 3 wt% | +28% | - | Enhanced stress transfer |
| Graphene/Epoxy [31] | 0.45 wt% | +31.3% | - | Homogeneous dispersion & interfacial adhesion |
| Graphene/PMMA [31] | - | - | +30% vs. CNT/PMMA | High surface area & 2D geometry |
| GnP/LCP [29] | 5 wt% | +21% | +32% | Filler alignment & strong interfacial interaction |
The Critical Role of Dispersion and Novel Fabrication Routes
Achieving homogeneous nanofiller dispersion is paramount, as agglomeration creates stress concentration points that compromise mechanical performance [31]. Genuine nanocomposites represent a paradigm shift by bypassing traditional dispersion challenges. This approach involves creating nanoscale reinforcement in situ during processing (e.g., by cold-drawing polymer blends to form nanofibrils) rather than blending pre-formed nanofillers. This method can potentially increase tensile strength and modulus by 300-400% compared to conventional techniques [24].
Diagram 2: This diagram outlines the primary mechanisms and critical factors responsible for mechanical property enhancement in polymer nanocomposites.
Experimental Protocol: Solution Casting for Graphene Nanocomposites
Objective: To fabricate exfoliated graphene nanoplatelet (GnP)/liquid crystalline polymer (LCP) nanocomposite films with enhanced mechanical properties via solution casting.
Materials:
- Matrix: Liquid crystalline polymer (e.g., Parmax, poly(benzoyl-1,4-phenylene)-co-(1,3-phenylene))
- Filler: Exfoliated graphite nanoplatelets (GnP-15, ~10-15 μm diameter)
- Solvent: Chloroform (suitable for the polymer and filler)
Equipment:
- Fume hood
- Ultrasonic bath and probe sonicator
- Vacuum oven
- Teflon-coated glass plates
- Universal testing machine (for tensile testing)
- Field Emission Scanning Electron Microscope (FESEM)
Procedure:
- Polymer Solution Preparation: Dissolve LCP pellets in chloroform (e.g., 2% w/v) using mechanical stirring at room temperature until a clear, homogeneous solution is obtained.
- Filler Dispersion: Separately, disperse the desired weight percentage (e.g., 1-5 wt%) of GnP in chloroform using a combination of magnetic stirring and probe sonication (e.g., 200-300 W, 15-20 minutes in an ice bath to prevent overheating).
- Solution Mixing: Combine the GnP dispersion with the LCP solution. Stir the mixture vigorously for 1-2 hours, followed by bath sonication for 30-60 minutes to ensure homogeneous dispersion.
- Film Casting: Pour the final mixture onto a clean, level Teflon-coated glass plate. Cover the plate loosely with an inverted glass funnel to control the solvent evaporation rate.
- Solvent Evaporation: Allow the solvent to evaporate slowly at room temperature for 12-24 hours, followed by further drying in a vacuum oven at 50-60°C for 6-12 hours to remove residual solvent.
- Post-Processing (Optional - Alignment): For enhanced alignment, the dried film can be subjected to a mechanical buffing process using a soft cloth under light pressure to induce shear, aligning the LCP chains and GnP platelets.
- Characterization: Perform tensile tests according to ASTM D638. Examine the fracture surface and filler dispersion using FESEM.
Key Parameters:
- Solvent choice is critical and must adequately wet both the polymer and filler.
- Controlled, slow solvent evaporation helps prevent filler agglomeration and film defects.
- The highly aromatic structure of LCPs promotes strong π-π interactions with the graphene basal plane, enhancing interfacial adhesion [29].
Thermal Property Enhancement
Thermal Conductivity Mechanisms
Enhancing thermal conductivity in typically insulating polymers relies on establishing continuous pathways for efficient phonon transport. The key strategies include:
- Formation of Thermal Percolation Networks: Similar to electrical conductivity, a interconnected network of high-thermal-conductivity fillers (e.g., graphene, BN, CNTs) provides a low-resistance path for heat flow [33].
- Reduction of Interfacial Thermal Resistance: The high surface area of nanofillers, if coupled with good interfacial adhesion, minimizes Kapitza resistance (phonon scattering at interfaces) [33].
- Alignment of Anisotropic Fillers: Orienting high-aspect-ratio fillers (e.g., CNTs, graphene) in the through-plane or in-plane direction can lead to directional thermal management, which is critical for applications like electronics cooling [33].
Table 3: Thermal Property Enhancement in Polymer Nanocomposites
| Nanocomposite System | Filler Content | Thermal Conductivity | Enhancement | Application Relevance |
|---|---|---|---|---|
| PLA/CNT [30] | 3 wt% | - | +15% | Thermal management in 3D printed parts |
| BN/Polysiloxane [28] | - | - | - | Thermally conductive electrical insulator |
| Polymer Nanocomposites [33] | Low loadings | Isotropic & Anisotropic | Significant | Battery thermal management, TIMs |
Application-Oriented Thermal Management
The enhancement of thermal conductivity is not merely a numerical improvement but is driven by application demands. Key application areas highlighted in research include wearable electronics, thermal interface materials (TIMs), battery thermal management, dielectric capacitors, and solar thermal energy storage [33]. For instance, in electronic devices, polymer nanocomposites can dissipate heat efficiently, preventing performance degradation and failure.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Polymer Nanocomposites Research
| Material Category | Specific Examples | Key Function/Property | Application Notes |
|---|---|---|---|
| Carbon Nanofillers | Carbon Black (CB) [28] | Low-cost, forms conductive networks | Requires high loading (~15-20 wt%) for percolation in composites. |
| Carbon Nanotubes (CNTs) [30] | High aspect ratio, excellent electrical & thermal conductivity | Tendency to agglomerate; requires dispersion strategies. | |
| Graphene Nanoplatelets (GnP) [29] | 2D structure, high modulus (~1 TPa), high conductivity | Large surface area promotes strong interfacial interaction. | |
| Polymer Matrices | Polysiloxane [28] | Insulating, flexible, crosslinkable | Suitable for electric field-assisted alignment studies. |
| Polylactic Acid (PLA) [30] | Biodegradable, thermoplastic | Common matrix for fused deposition modeling (FDM) 3D printing. | |
| Liquid Crystalline Polymer (LCP) [29] | Inherently high strength and stiffness, can be aligned | Aromatic structure promotes π-π interaction with graphene. | |
| Specialty Additives | Boron Nitride (BN) [28] | Thermally conductive but electrically insulating | Ideal for thermal management where electrical insulation is critical. |
| Silica Nanoparticles [7] | Improves piezoelectric-elastic response | Used as secondary filler in multifunctional composites. |
This application note has detailed the principal mechanisms and methodologies for enhancing the electrical, mechanical, and thermal properties of polymer nanocomposites. The key to success lies in the strategic selection of nanofillers, the implementation of advanced processing techniques—such as external field alignment and solution casting that ensure optimal dispersion and distribution—and the thoughtful design of the polymer-filler interface. The provided protocols, data, and visual guides serve as a foundation for researchers to develop next-generation polymer nanocomposites with tailored multifunctional properties, pushing the boundaries of their application in advanced technological fields. Future research directions should continue to focus on overcoming dispersion challenges, precisely controlling nanofiller orientation in three dimensions, and achieving the synergistic enhancement of multiple properties simultaneously.
Synthesis Techniques and Transformative Biomedical Applications of Polymer Nanocomposites
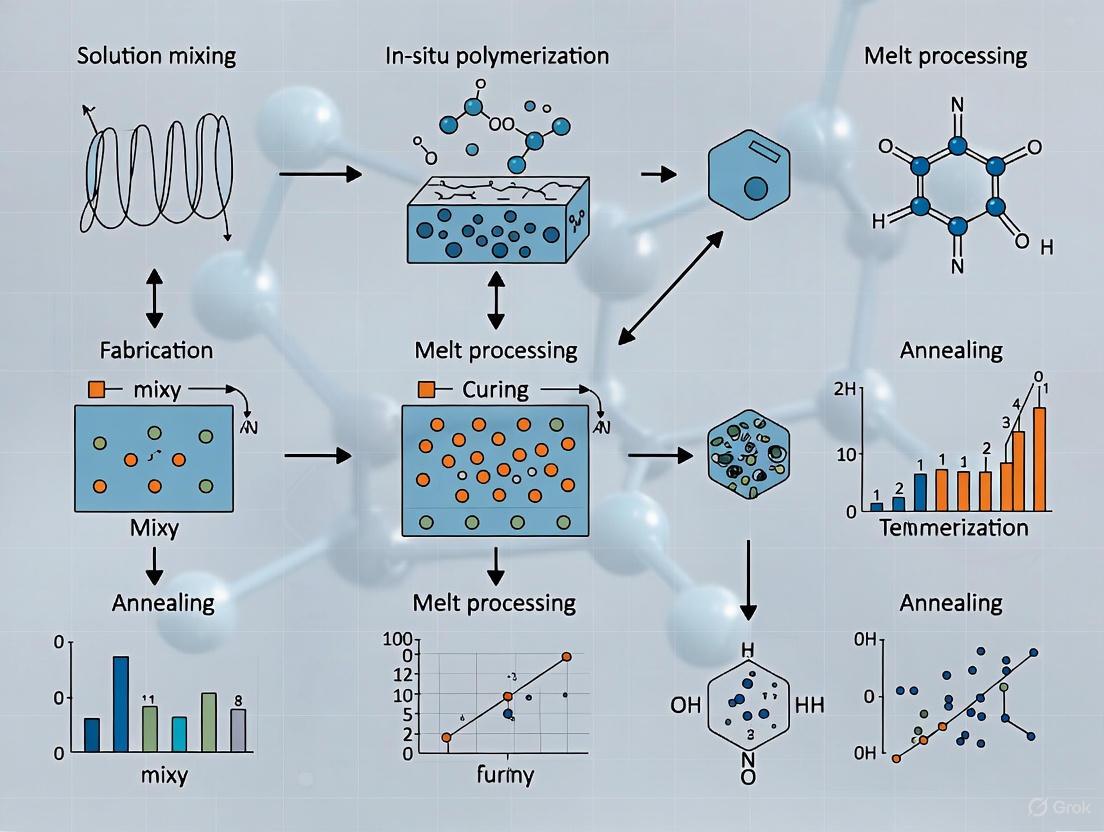
Polymer nanocomposites (PNCs) represent a advanced class of materials formed by dispersing nanoscale fillers within a polymer matrix, leading to significant enhancements in mechanical, thermal, electrical, and barrier properties compared to conventional polymers or microcomposites [4]. The fabrication methodology critically determines the dispersion state of nanoparticles and the resulting material performance. Achieving optimal nanoparticle dispersion remains a fundamental challenge due to thermodynamic drivers for agglomeration, which can cause conventional blending to yield microcomposite-like behavior rather than true nanocomposite performance [24] [34]. This application note details three core fabrication methodologies—in-situ polymerization, solution blending, and melt compounding—within the context of advanced materials research, providing structured protocols and analytical frameworks for researchers and drug development professionals engaged in nanocomposite development.
Comparative Analysis of Fabrication Methods
Table 1: Comparative analysis of core fabrication methodologies for polymer nanocomposites
| Parameter | In-Situ Polymerization | Solution Blending | Melt Compounding |
|---|---|---|---|
| Core Principle | Nanoparticles dispersed in monomer followed by polymerization [35] [36] | Nanoparticles and polymer dissolved/dispersed in solvent followed by solvent removal [36] | Nanoparticles mixed with polymer melt using high-shear equipment [37] [36] |
| Dispersion Quality | Generally good; particles nucleate on active polymer sites [36] | Good with optimal solvent selection [36] | Variable; dependent on shear forces and compatibility [38] [37] |
| Key Advantages | Strong nanoparticle-polymer adhesion [36]; applicable to complex shapes [35] | Simple process [36]; good for laboratory-scale research [39] | Solvent-free [36]; industrially scalable [37]; compatible with standard polymer processing |
| Key Limitations | Viscosity increase during process [36]; potential polymerization interference | Solvent toxicity and removal challenges [36]; environmental concerns | High-temperature exposure risk [36]; potential nanoparticle degradation/agglomeration |
| Industrial Scalability | Moderate | Moderate (solvent management challenges) | High |
| Energy Consumption | Moderate | High (due to solvent removal) | Moderate to High |
| Common Applications | Conductive composites [40]; antimicrobial textiles [36]; biomedical devices [4] | Membrane technology [36]; sensor materials; specialty coatings | Automotive components [4] [40]; packaging materials [4]; structural composites |
Table 2: Property enhancement ranges achieved through different methodologies
| Property Enhancement | In-Situ Polymerization | Solution Blending | Melt Compounding |
|---|---|---|---|
| Tensile Strength Increase | Up to 300-400% with optimal systems [24] | 50-200% depending on filler dispersion [39] | 30-150% dependent on compounding parameters [37] |
| Electrical Conductivity | Can achieve conductive networks at low percolation [40] | Good for creating conductive pathways [40] | Conductive with sufficient shear and dispersion [40] |
| Thermal Stability | Significant improvement [4] | Moderate to significant improvement [39] | Moderate improvement [4] |
| Barrier Properties | Good enhancement reported [4] | Excellent enhancement possible [4] | Good enhancement for packaging [4] |
Methodological Deep Dive
In-Situ Polymerization
In-situ polymerization involves dispersing nanoparticles within a monomer or monomer solution followed by polymerization, enabling the growth of polymer chains in the presence of nanofillers. This method often results in excellent filler dispersion and strong polymer-filler interfacial adhesion because nanoparticles can nucleate and grow on active sites of the macromolecular chains [36]. This approach is particularly valuable for creating nanocomposites with metals (silver, gold), carbon-based nanomaterials, and other functional fillers for advanced applications.
Experimental Protocol: In-Situ Polymerization of Nylon 6/MWCNT Nanocomposites
Research Reagent Solutions:
- Multi-Walled Carbon Nanotubes (MWCNTs): Provide reinforcement and electrical conductivity [40].
- ε-Caprolactam Monomer: Precursor for nylon 6 polymerization [36].
- Polymerization Initiator/Catalyst: (e.g., anionic catalyst for ring-opening polymerization) - Facilitates the polymerization reaction [36].
- Compatibilizer/Surfactant: (e.g., polyvinylpyrrolidone - PVP) - Aids in nanotube dispersion and prevents agglomeration [36].
Equipment: Ultrasonic bath or probe sonicator, three-neck reaction flask equipped with mechanical stirrer, nitrogen purge system, temperature-controlled oil bath, vacuum oven.
Step-by-Step Procedure:
- Nanofiller Pre-dispersion: Suspend MWCNTs (e.g., 1-5 wt%) in the liquid ε-caprolactam monomer. Subject the mixture to high-intensity probe sonication for 30-60 minutes under a nitrogen atmosphere to achieve a homogeneous dispersion.
- Reactor Setup: Transfer the dispersion to a clean, dry three-neck flask equipped with a mechanical stirrer and nitrogen inlet.
- In-Situ Polymerization: Heat the mixture to the polymerization temperature (e.g., ~250°C for anionic polymerization of nylon 6) with continuous stirring. Add the catalyst system to initiate the ring-opening polymerization of ε-caprolactam. Maintain reaction conditions for 2-4 hours.
- Product Recovery: After completion, allow the reaction mass to cool. Crush the resulting solid and purify by washing with appropriate solvents to remove residual monomer and catalyst.
- Post-Processing: Dry the purified nanocomposite in a vacuum oven at 80°C for 24 hours. The resulting material can be processed via melt extrusion for pelletization or direct molding [36] [40].
Solution Blending
Solution blending relies on dispersing nanoparticles and dissolving the polymer matrix in a suitable solvent, followed by mixing and subsequent solvent removal to form the nanocomposite. This method is particularly effective for achieving good nanoparticle dispersion with minimal agglomeration, making it ideal for laboratory-scale research and applications requiring high optical clarity or specific interfacial properties.
Experimental Protocol: Solution Blending for PVDF-HFP/Clay Nanocomposites
Research Reagent Solutions:
- Poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP) Matrix: Provides piezoelectric properties and flexibility [36].
- Organically Modified Nanoclays (e.g., Montmorillonite): Enhance mechanical strength and dielectric properties [36].
- Solvent: N,N-Dimethylformamide (DMF) - Effectively dissolves PVDF-HFP and disperses nanoclays [36].
- Non-Solvent: Deionized Water or Methanol - Acts as an anti-solvent for coagulation [36].
Equipment: Magnetic stirrer/hotplate, ultrasonic bath or sonicator, solvent-resistant containers, coagulation bath, vacuum filtration setup, vacuum oven.
Step-by-Step Procedure:
- Polymer Solution Preparation: Dissolve PVDF-HFP pellets (e.g., 10-15% w/v) in DMF with continuous stirring at 50-60°C until a clear, homogeneous solution is obtained.
- Nanofiller Dispersion: Separately, disperse the desired weight percentage (e.g., 1-7 wt%) of nanoclays in DMF using ultrasonication for 30-45 minutes to exfoliate the clay layers.
- Blending: Gradually add the nanoclay dispersion to the polymer solution with vigorous stirring. Continue stirring for several hours (e.g., 4-6 h) to ensure homogeneous mixing.
- Solvent Removal & Coagulation: Pour the blended solution slowly into a large excess of vigorously stirred non-solvent (e.g., water or methanol). This causes the polymer nanocomposite to coagulate and precipitate.
- Washing and Drying: Collect the precipitated nanocomposite by vacuum filtration. Wash thoroughly with fresh non-solvent to remove residual DMF. Dry the final product in a vacuum oven at 60-80°C for at least 24 hours until constant weight is achieved [36].
Melt Compounding
Melt compounding involves the mechanical mixing of nanoparticles with a polymer melt using high-shear equipment, typically twin-screw extruders. This solvent-free process is highly attractive for industrial-scale production due to its compatibility with existing polymer processing infrastructure, environmental friendliness, and high efficiency.
Experimental Protocol: Melt Compounding of PA6/Organoclay Nanocomposites
Research Reagent Solutions:
- Polyamide 6 (PA6) Matrix: Engineering thermoplastic with good mechanical properties.
- Organically Modified Clay (e.g., Cloisite 15A): Swells and exfoliates under shear to enhance properties [37].
- Compatibilizer: Maleic anhydride-grafted polypropylene (PP-g-MA) - Improves interfacial adhesion in non-polar systems (optional for PA6) [37] [40].
Equipment: Twin-screw extruder, drying oven, gravimetric feeders, water bath, pelletizer.
Step-by-Step Procedure:
- Material Pre-conditioning: Dry PA6 pellets and organoclay in a vacuum oven at 80°C for 12 hours to remove moisture.
- Feeding: Pre-mix the dried PA6 pellets with the target weight percentage (e.g., 2-5 wt%) of organoclay using a tumble blender. For polyolefin matrices, include 2-5% compatibilizer.
- Melt Compounding: Feed the pre-mix into a twin-screw extruder using a gravimetric feeder. Set appropriate temperature profiles along the extruder barrels (e.g., 230-260°C for PA6) and screw speed (e.g., 200-300 rpm) to achieve optimal shear mixing.
- Strand Pelletizing: Extrude the melt through a strand die, cool the strands in a water bath, and pelletize using a rotary cutter.
- Post-Processing: Dry the pellets again before subsequent processing steps like injection molding or compression molding [37].
Advanced Considerations and Emerging Trends
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential materials for polymer nanocomposite fabrication
| Material Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| Nanofillers | Carbon nanotubes (CNTs), Graphene, Nanoclays, Silver nanoparticles | Impart enhanced mechanical, electrical, thermal, or antimicrobial properties [4] [36] [40] | Surface energy and aspect ratio critically influence dispersion and percolation threshold [40] |
| Polymers | Polyamide (PA), Polypropylene (PP), Poly(vinylidene fluoride) (PVDF) | Act as the continuous matrix material | Polarity and functional groups affect nanoparticle compatibility |
| Compatibilizers | Maleic anhydride-grafted polymers (e.g., PP-g-MA) | Improve interfacial adhesion between filler and matrix [40] | Essential for non-polar polymers with polar nanofillers |
| Surfactants/Stabilizers | Polyvinylpyrrolidone (PVP) | Prevent nanoparticle agglomeration in solutions and melts [36] | Used in in-situ and solution methods; choice depends on nanoparticle surface chemistry |
| Solvents | N,N-Dimethylformamide (DMF), Toluene, Chloroform | Dissolve polymer and disperse nanoparticles in solution blending [36] | Selection based on solubility parameters, boiling point, and toxicity |
Advanced Methodological Frameworks
Future developments are increasingly focused on overcoming dispersion limitations through "create rather than add" approaches, such as in-situ generation of nanoparticles within polymer matrices [24] [36]. This strategy skips the thermodynamically challenging dispersion step entirely, instead creating nanoreinforcements during composite manufacturing. Examples include the cold drawing of polymer blends to generate nanofibrils or the chemical transformation of precursors directly within the polymeric host [24] [36]. Furthermore, advanced manufacturing techniques like 3D printing are being integrated with nanocomposite fabrication, enabling the creation of complex, multifunctional architectures with precise nanofiller placement for applications in biomedicine, soft robotics, and electronics [35].
The convergence of electrospinning and 3D/4D printing represents a transformative advancement in the fabrication of polymer nanocomposites with complex architectures. While electrospinning enables the production of nano- to micro-scale fibrous structures that closely mimic the native extracellular matrix, it often results in scaffolds with insufficient mechanical properties for structurally complex tissues [41]. Conversely, 3D printing offers unparalleled geometrical freedom and control over macroscale architecture but typically struggles to achieve the nanoscale resolution required for optimal cellular interaction [41]. The synergy between these technologies creates a powerful platform for developing multi-scale hierarchical structures that overcome the individual limitations of each technique [42] [43].
This integration is particularly significant for biomedical applications, where the structural and functional complexity of native tissues demands sophisticated manufacturing approaches. The combination of electrospun nanofibers with 3D printed elements produces composite structures with superior properties, including enhanced mechanical strength, tailored porosity, and biomimetic topography that directs cellular behavior [42] [43] [44]. Furthermore, the emergence of 4D printing—which introduces the dimension of time through stimuli-responsive materials—adds dynamic functionality to these architectures, enabling shape-morphing behaviors or controlled degradation profiles that better replicate physiological processes [45].
Fundamental Principles and Technologies
Electrospinning Fundamentals
Electrospinning is a nanofabrication technique that utilizes high electric voltages to produce polymeric fibers with diameters ranging from micro- to nanometers [44]. The process involves applying a strong electric field between a polymer solution or melt contained in a spinneret (typically a metallic needle connected to a syringe) and a conductive collector. When the electrostatic forces overcome the surface tension of the polymer solution, a Taylor cone forms at the needle tip, and a charged polymer jet is ejected toward the collector [41]. This jet undergoes stretching and whipping instabilities during its trajectory, leading to extreme fiber thinning and eventual solidification through solvent evaporation or cooling, resulting in the deposition of solid nanofibers on the collector [41] [44].
Critical Parameters Influencing Electrospinning:
Solution and Polymer Parameters: Polymer molecular weight and concentration directly affect solution viscosity, which determines fiber morphology. Lower molecular weights or concentrations often result in bead formation instead of continuous fibers, while excessively high values can cause clogging [41]. Solvent choice impacts solution conductivity, dielectric constant, and evaporation rate, all influencing final fiber diameter and morphology [41].
Process Parameters: Applied voltage (typically 10-25 kV) controls the electrostatic forces driving fiber formation and stretching [41]. The tip-to-collector distance affects jet flight time and solvent evaporation rate, with greater distances generally producing thinner, more uniform fibers [41]. Flow rate determines solution volume available for electrospinning, with lower rates typically yielding thinner fibers [41].
3D/4D Printing Technologies
3D printing, or additive manufacturing (AM), encompasses a suite of technologies that build three-dimensional objects layer-by-layer from digital models, offering unrivalled geometrical freedom and customization capabilities [44]. In the context of polymer nanocomposites, extrusion-based methods like fused filament fabrication (FFF) are particularly relevant, where thermoplastic filaments are melted and deposited through a nozzle according to computer-controlled paths [46]. These techniques allow the fabrication of highly complex components with tailored architectures but are generally considered "low-resolution" techniques compared to electrospinning, with limited ability to manipulate structural details at the submicrometric scale [41].
4D printing represents an advanced evolution of 3D printing, where the fabricated structures can change their shape, properties, or functionality over time in response to external stimuli such as temperature, moisture, light, or magnetic fields [45]. This dynamic behavior is achieved through the use of smart materials, including shape-memory polymers, hydrogels, and stimuli-responsive composites. When combined with electrospun nanofibers, 4D printing enables the creation of sophisticated structures that can undergo programmed morphological changes, offering exciting possibilities for advanced biomedical applications such as self-fitting implants or programmable drug delivery systems [45].
Integrated Fabrication Approaches and Quantitative Analysis
Hybrid Fabrication Strategies
The integration of electrospinning and 3D/4D printing has primarily been realized through two distinct fabrication approaches, each offering unique advantages for specific applications.
Multi-Layered Architectures: This approach involves the sequential or alternating deposition of electrospun nanofibers and 3D printed elements to create composite scaffolds with multi-scale hierarchical organization [43] [44]. In one implementation, a 3D printed framework provides structural support and macroscopic geometry, while electrospun nanofibers integrated between printed layers enhance biomimetic topography and cellular interaction [41] [44]. This strategy has been successfully applied to tissue engineering scaffolds, where the 3D printed components maintain structural integrity while the electrospun layers improve cell seeding efficiency and tissue formation [44]. For instance, primary bovine articular chondrocyte entrapment and extracellular matrix production were significantly enhanced in alternating layers of electrospun nanofibers and 3D printed meshes compared to single-technology constructs [44].
Fiber-Reinforced Composite Inks: This approach incorporates electrospun nanofibers directly into 3D printing inks prior to the printing process, creating composite materials with enhanced mechanical properties and functionality [43] [44]. The nanofibers act as reinforcement within the printed matrix, improving tensile strength, storage modulus, and other mechanical characteristics while maintaining printability [46] [43]. This method overcomes challenges associated with poor dispersion of conventional nanoscale additives in polymer melts and the difficult processability of resulting high-viscosity materials [46]. A notable example includes the fabrication of poly(lactide acid) (PLA) nanocomposites with electrospun nanofiber interleaves, where systematic variation of nanofiber content demonstrated significant improvements in mechanical properties [46].
Table 1: Comparative Analysis of Hybrid Fabrication Approaches
| Parameter | Multi-Layered Architecture | Fiber-Reinforced Composite Inks |
|---|---|---|
| Fabrication Sequence | Sequential deposition of electrospun mats and 3D printed elements | Pre-incorporation of fibers into printing ink before fabrication |
| Mechanical Reinforcement | Through interlayer adhesion and distribution | Through homogeneous dispersion within matrix |
| Structural Control | Macroscopic (3D printing) and microscopic (electrospinning) | Primarily macroscopic with enhanced bulk properties |
| Material Compatibility | Can combine disparate materials in discrete layers | Requires compatibility between fibers and matrix material |
| Key Advantages | Independent optimization of each layer, high customizability | Improved mechanical properties, simplified fabrication process |
| Limitations | Potential delamination, complex process optimization | Potential nozzle clogging, fiber alignment challenges |
| Typical Applications | Tissue engineering scaffolds, filtration systems | Enhanced functional composites, reinforced structures |
Quantitative Performance Data
The synergistic combination of electrospinning and 3D printing yields measurable improvements in structural and functional properties compared to individual technologies.
Table 2: Quantitative Performance Enhancement in Hybrid Structures
| Property Metric | 3D Printing Alone | Electrospinning Alone | Combined Technology | Application Context |
|---|---|---|---|---|
| Tensile Strength | Baseline | Variable | Significant improvement with nanofiber integration [46] | PLA nanocomposites [46] |
| Compressive Modulus | ~23 kPa | Not typically measured | ~89 kPa (4-fold increase) [43] | Alginate-PCL scaffolds [43] |
| Elastic Recovery | Up to 30% strain | Not typically measured | Up to 45% strain [43] | Alginate-PCL scaffolds [43] |
| Cell Seeding Efficiency | ~30% | Not applicable | ~80% with high-density electrospun mats [44] | PCL scaffolds with hADSCs [44] |
| Surface Area | Limited by print resolution | High (specific to nanofibers) | Combines macroscopic and nanoscale features | Tissue engineering scaffolds [41] |
| Pore Size Control | Macroscopic (hundreds of µm) | Microscopic (submicron to few µm) | Multi-scale hierarchical porosity | Biomimetic scaffolds [41] |
Experimental Protocols and Methodologies
Protocol 1: Fabrication of Multi-Layered Scaffolds with Alternating Electrospun and 3D Printed Layers
Application Context: This protocol describes the fabrication of composite scaffolds for tissue engineering applications, particularly suited for bone, cartilage, or skin tissue regeneration where both structural integrity and enhanced cellular interaction are required [41] [44].
Materials and Equipment:
- 3D Printer (Fused Filament Fabrication system)
- Electrospinning apparatus with high-voltage power supply
- Poly(lactic acid) (PLA) or poly(ε-caprolactone) (PCL) filament for 3D printing
- Polymer solution for electrospinning (e.g., PCL in appropriate solvent)
- Solvent system (e.g., dimethylformamide, tetrahydrofuran, or chloroform)
- Syringe and metallic needle for electrospinning
- Collectors (flat plate or rotating mandrel)
Procedure:
- 3D Printing of Primary Framework:
- Design a 3D model with desired macro-architecture using CAD software, typically featuring a 0°/90° laydown pattern with square pores (e.g., 350 µm) to facilitate cellular infiltration [44].
- Convert the model to G-code and set printing parameters: nozzle temperature 190-220°C (material-dependent), bed temperature 60°C, layer thickness 0.2 mm, printing speed 40-60 mm/s [47].
- Execute the printing process to create the primary structural framework.
Electrospinning Solution Preparation:
- Prepare polymer solution by dissolving PCL in an appropriate solvent system (e.g., 10-15% w/v) with continuous stirring for 6-12 hours until complete dissolution [41].
- Adjust solution parameters based on desired fiber morphology: higher concentrations (12-16%) for larger fiber diameters, lower concentrations (8-12%) for finer fibers [41].
Integration of Electrospun Layers:
- Mount the 3D printed structure on the electrospinning collector, ensuring secure fixation.
- Set electrospinning parameters: applied voltage 15-25 kV, flow rate 0.5-2 mL/h, tip-to-collector distance 10-20 cm, relative humidity 30-50% [41] [44].
- For aligned fiber deposition, use a rotating mandrel collector with controlled speed (e.g., 1000-3000 rpm) [44].
- Deposit electrospun fibers for a predetermined duration (45-360 seconds) to achieve desired nanofiber density [44].
- Alternate between 3D printing and electrospinning steps to build multi-layered architectures.
Post-Processing:
- Vacuum-dry the composite structure for 24 hours to remove residual solvents.
- Characterize the resulting scaffold morphology, mechanical properties, and cellular response.
Protocol 2: Nanofiber Interleaving for Enhanced Mechanical Properties
Application Context: This protocol details the incorporation of electrospun nanofiber mats as interleaves within 3D printed structures to enhance mechanical properties, particularly addressing the limited polymer strength in fused filament fabrication due to high cooling rates and lack of crystallinity [46].
Materials and Equipment:
- Fused filament fabrication 3D printer
- Electrospinning apparatus
- Poly(lactide acid) (PLA) filament for printing
- PLA or other compatible polymer for electrospinning
- Appropriate solvents
- Cutting tools for nanofiber mat precision shaping
Procedure:
- Electrospun Nanofiber Mat Fabrication:
- Prepare polymer solution for electrospinning (e.g., 16% polyacrylonitrile in dimethyl sulfoxide) with continuous stirring until complete dissolution [47].
- Set electrospinning parameters: voltage 80 kV, carriage speed 100 mm/s, substrate distance 240 mm, temperature 22-23°C, relative humidity 32% [47].
- Collect nanofiber mats on appropriate substrate (e.g., polypropylene) for 8 minutes to achieve areal weight of 1.7-2.1 g/m² [47].
Precision Shaping of Nanofiber Interleaves:
- Carefully remove nanofiber mats from collection substrate.
- Cut into precise shapes matching the geometry of 3D printed layers using laser cutting or precision blades.
Sequential 3D Printing with Interleave Integration:
- Begin 3D printing of initial layers using standard parameters (nozzle temperature 190°C for PLA, bed temperature 60°C, layer thickness 0.2 mm) [47].
- Pause printing after completion of designated layers.
- Manually place pre-cut nanofiber interleave on top of the last printed layer.
- Resume printing process, ensuring proper nozzle temperature to promote adhesion between printed layers and nanofiber interleaves [46].
- Repeat sequence as needed to create multiple interleaved regions throughout the structure.
Adhesion Optimization:
Characterization:
- Evaluate mechanical properties through tensile testing and dynamic mechanical analysis.
- Characterize morphology and interface quality using scanning electron microscopy.
- Assess storage modulus and tensile strength improvements compared to non-interleaved structures [46].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for Hybrid Electrospinning-3D Printing Research
| Material/Reagent | Function/Application | Key Considerations | Representative Examples |
|---|---|---|---|
| Poly(lactic acid) (PLA) | Biodegradable polymer for both electrospinning and 3D printing | Good mechanical properties, tunable degradation rate | 3D printing filament, electrospinning solutions [46] [47] |
| Poly(ε-caprolactone) (PCL) | Semi-crystalline biodegradable polymer | Slower degradation rate, flexibility, FDA approval for certain devices | Tissue engineering scaffolds, flexible structures [43] [44] |
| Polyacrylonitrile (PAN) | Synthetic polymer for electrospun nanofibers | High strength, chemical resistance, can be carbonized | Filtration applications, composite reinforcement [47] |
| Dimethyl Sulfoxide (DMSO) | Solvent for electrospinning | Polar aprotic solvent, moderate toxicity | Dissolving PAN and other polymers [47] |
| Thermoplastic Polyurethane (TPU) | Flexible polymer for 3D printing | Elasticity, abrasion resistance, strong adhesion to electrospun fibers | Flexible composites, filtration systems [47] |
| PEDOT:PSS | Conductive polymer complex | Aqueous dispersion, forms conductive films | Coating 3D printed objects to modify electrical properties [47] |
| Alginate | Natural polysaccharide for bioinks | Biocompatibility, ionic crosslinking, often combined with electrospun fibers | Bioinks for extrusion-based bioprinting [43] |
Advanced Applications and Technology Implementation
Biomedical Applications
Tissue Engineering Scaffolds: The integration of electrospinning and 3D printing has shown remarkable success in creating tissue-specific scaffolds for various applications. For bone tissue engineering, screw-assisted extrusion-based additive manufacturing combined with electrospun PCL nanofibers has demonstrated enhanced human adipose-derived stem cell (hADSC) attachment and osteogenic differentiation [44]. Scaffolds with high-density electrospun mats exhibited cell seeding efficiencies of approximately 80%, significantly higher than scaffolds without nanofibers (~30%) [44]. For skin tissue regeneration, multi-layer substitutes have been developed by depositing electrospun PCL and keratin fibers onto both surfaces of 3D printed PCL scaffolds, successfully mimicking the histological structure of skin [44]. The top electrospun layer (100 µm thick, ~0.7 µm fiber diameter) supported human immortalized keratinocytes (HaCaT), while the bottom layer (300 µm thick, ~1.7 µm fiber diameter) enabled proliferation of normal human dermal fibroblasts (NHDF) [44].
Intervertebral Disc Regeneration: Complex scaffolds addressing intervertebral disc degeneration represent a sophisticated application of hybrid technology [44]. These constructs incorporate three main elements: a 3D printed PLA frame simulating the disc structure; bundles of oriented porous nanofibers of poly(l-lactide)/POSS mimicking the annulus fibrosus; and a gellan gum/poly(ethylene glycol) diacrylate hydrogel encapsulating bone marrow mesenchymal stem cells reproducing the nucleus pulposus [44]. Animal implantation tests demonstrated effectiveness comparable to reimplanted autologous discs in maintaining disc height and producing new extracellular matrix components including proteoglycans and collagen [44].
Filtration and Environmental Applications
The combination of electrospinning and 3D printing enables the fabrication of advanced filtration systems with enhanced mechanical stability and functionality [47]. Electrospun nanofiber mats provide exceptional filtration capabilities due to their small pore sizes and high surface-to-volume ratio, allowing for filtering of minute particles or molecules [47]. However, these nanofiber mats are prone to mechanical damage, limiting their practical application. Integration with 3D printed scaffolds addresses this limitation, creating mechanically stable filters with nanofibrous surfaces [47].
Research has demonstrated that electrospinning directly onto 3D printed thermoplastic polyurethane (TPU) objects results in strong adhesion between the materials, creating composites suitable for filtration applications [47]. In contrast, adhesion to common rigid printing polymers like PLA is generally insufficient without additional surface modifications or adhesive layers [47]. This approach paves the way for developing mechanically robust filters with the enhanced surface functionality provided by nanofibers, potentially incorporating materials that enable additional treatment of filtered substances through photo-degradation or antimicrobial properties [47].
Future Perspectives and Emerging Trends
The integration of electrospinning with 3D/4D printing continues to evolve with several emerging trends shaping its future development. The incorporation of artificial intelligence (AI) and machine learning for process optimization and material design represents a significant advancement, enabling predictive modeling of process parameters and their effects on final structure properties [35]. Additionally, the development of nanoscale additive manufacturing techniques promises to further bridge the resolution gap between conventional 3D printing and electrospinning [35].
The concept of 5D printing (3D + time + information) expands upon 4D printing by incorporating real-time data and feedback systems, creating structures that can adapt dynamically to their environment [35]. Furthermore, sustainable closed-loop recycling systems for multi-material nanocomposites address growing environmental concerns [35]. As these technologies mature, their convergence is expected to enable increasingly sophisticated architectures with programmed functionality across multiple length scales, opening new possibilities in advanced applications ranging from personalized medical implants to adaptive soft robotics and smart filtration systems.
Application Note: Covalent Surface Modification of Inorganic Nanoparticles
The enhancement of material properties in polymer nanocomposites is highly dependent on the interface between the inorganic filler and the polymeric matrix. Covalent surface modification represents a foundational approach to tailor this particle-matrix interface, significantly improving dispersion homogeneity and interfacial adhesion [48]. This application note details a two-step modification strategy for zirconia (ZrO₂) nanoparticles, enabling the covalent attachment of polymerizable organic moieties to the particle surface. This "grafting-through" technique facilitates strong covalent integration of the nanoparticles into a polystyrene matrix during polymerization, leading to substantial improvements in mechanical properties [48].
Detailed Experimental Protocol
Materials:
- Zirconia (ZrO₂) nanoparticles (synthesized via non-aqueous sol-gel method)
- 3-Aminopropyltriethoxysilane (APTES, 99%)
- N,N'-Diisopropylcarbodiimide (DIC, 98%)
- Ligands: 10-Undecenoic acid (UD, 98%), Undecanoic acid (UND, 98%), 4-Vinylbenzoic acid (vBA, 97%), Benzoic acid (BA, 99.5%)
- Solvents: Ethanol, ethyl acetate (technical grade)
- Monomeric styrene, peroxide initiator (e.g., CUROX M-312)
Procedure:
- First Step: Silanization with APTES
- Begin with a suspension of as-synthesized ZrO₂ nanoparticles in benzyl alcohol.
- Add APTES (45 wt% relative to theoretical ZrO₂ content) to the nanoparticle suspension.
- Heat the mixture under reflux at 110 °C with stirring for 12 hours.
- Precipitate the APTES-modified nanoparticles (ZrO₂-APTES) by adding ethyl acetate.
- Separate the particles via centrifugation (6280 rcf for 10 minutes).
- Redisperse the precipitate in ethanol, reprecipitate with ethyl acetate, and repeat this washing cycle three times to remove excess APTES and by-products.
- Store the final ZrO₂-APTES nanoparticles in ethanol.
Second Step: Ligand Coupling via Carbodiimide Chemistry
- Disperse the ZrO₂-APTES nanoparticles in ethanol at a concentration of 0.01 g/mL and sonicate for 20 minutes.
- Prepare a separate mixture of the desired carboxylic acid ligand (e.g., UD, UND, vBA, BA) and DIC in a 1:1.5 molar ratio in ethanol.
- Add the ligand/DIC mixture to the ZrO₂-APTES dispersion and stir at room temperature for 12 hours.
- Centrifuge the suspension to recover the modified nanoparticles.
- Wash the particles thoroughly with ethanol and ethyl acetate to remove any unreacted reagents.
- Dry the final functionalized nanoparticles (e.g., ZrO₂-vBA, ZrO₂-UD) under vacuum.
Nanocomposite Fabrication and Testing
- Incorporate the modified ZrO₂ nanoparticles into monomeric styrene at a defined loading (e.g., 3 wt%).
- Add a peroxide initiator and polymerize the mixture.
- Characterize the mechanical properties of the resulting nanocomposite, specifically measuring the Young's modulus via standardized indentation tests (e.g., DIN EN ISO 2439) [48].
Results and Data Analysis
The covalent linkage of polymerizable ligands to the nanoparticle surface leads to a marked enhancement in the mechanical properties of the resulting polystyrene nanocomposite.
Table 1: Mechanical Enhancement from Covalent Surface Modification
| Nanoparticle Type | Surface Ligand | Ligand Functionality | Young's Modulus Increase |
|---|---|---|---|
| ZrO₂-vBA | 4-Vinylbenzoic acid | Polymerizable (Aromatic vinyl) | Up to 28% at 3 wt% loading |
| ZrO₂-UD | 10-Undecenoic acid | Polymerizable (Aliphatic vinyl) | Significant improvement |
| ZrO₂-BA | Benzoic acid | Inert (Aromatic) | Lower improvement |
| ZrO₂-UND | Undecanoic acid | Inert (Aliphatic) | Lower improvement |
The data demonstrates that nanoparticles functionalized with terminal vinyl groups (ZrO₂-vBA, ZrO₂-UD) that can copolymerize with the styrene matrix provide superior reinforcement compared to those with inert ligands, underscoring the critical role of a covalent particle-matrix linkage [48].
Workflow Visualization
Application Note: Wet Chemical Surface Functionalization of Polymer Scaffolds
Surface functionality is a critical determinant of biological response for polymeric biomaterials used in tissue engineering and drug delivery [49]. Wet chemical techniques, such as hydrolysis and aminolysis, are straightforward and effective methods for introducing specific functional groups (e.g., -COOH, -NH₂) onto the surfaces of polymer fibers and films. These functional groups alter surface hydrophilicity and serve as anchor points for further conjugation of bioactive molecules, thereby improving cell adhesion, migration, and overall biocompatibility [49].
Detailed Experimental Protocols
Protocol 2.2.1: Alkaline Hydrolysis of PLGA Scaffolds
- Objective: To generate carboxylate (-COO⁻) groups on the surface of poly(lactide-co-glycolide) (PLGA) scaffolds.
- Materials: PLGA scaffold, Sodium Hydroxide (NaOH) solution (0.01 N), N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC), N-Hydroxysuccinimide (NHS), Buffer (pH = 6), Deionized (DI) Water.
- Procedure:
- Immerse freeze-dried PLGA scaffolds in 0.01 N NaOH solution.
- Incubate for 30 minutes at 25°C under gentle agitation.
- Rinse the hydrolyzed scaffolds thoroughly with DI water three times to remove residual NaOH.
- For subsequent conjugation, activate the newly formed carboxyl groups by immersing the scaffolds in a solution containing EDC (40 mM) and NHS (80 mM) in pH 6 buffer.
- Keep the reaction at 4°C for 4 hours [49].
Protocol 2.2.2: Aminolysis of PCL Films
- Objective: To introduce primary amine (-NH₂) groups onto the surface of poly(ε-caprolactone) (PCL) films.
- Materials: PCL films, 1,6-Hexanediamine, Isopropanol.
- Procedure:
- Prepare an aminolysis solution by dissolving 1,6-hexanediamine in isopropanol (e.g., 6% w/v).
- Immerse the PCL films in the aminolysis solution, ensuring complete coverage.
- Heat the solution to 50°C and incubate for a defined duration (e.g., 1-15 minutes).
- After treatment, rinse the aminolyzed PCL films extensively with Milli-Q water for 4 hours at room temperature to remove any free diamine [49] [50].
Results and Data Analysis
The efficiency of wet chemical treatments depends on polymer type, solvent, and reaction time. For example, the rate of PCL aminolysis is significantly higher in isopropanol than in water due to better wetting of the hydrophobic polymer surface [50]. Furthermore, susceptibility to aminolysis varies among aliphatic polyesters, generally following the order: PLLA > PLCL > PCL [51].
Table 2: Wet Chemical Surface Modification of Polymers
| Polymer | Technique | Reagent Conditions | Introduced Functionality | Primary Application |
|---|---|---|---|---|
| PLGA | Alkaline Hydrolysis | 0.01 N NaOH, 30 min, 25°C | Carboxyl (-COOH) | Conjugation of bioactive peptides (e.g., RGD) for enhanced cell adhesion [49] |
| PCL | Aminolysis | 1,6-Hexanediamine/Isopropanol, 50°C, 1-15 min | Primary Amine (-NH₂) | Immobilization of proteins (e.g., collagen, gelatin) to create a bioactive surface [49] [52] |
| PET | Alkaline Hydrolysis | 4 M NaOH, 70°C, 3h | Carboxyl (-COOH) | Significant improvement of surface hydrophilicity for improved biocompatibility [49] |
| PES | Aminolysis | 10 wt% Diethylenetriamine (DETA) in Water | Primary Amine (-NH₂) | Subsequent immobilization of carboxymethyl cellulose (CMC) [49] |
Workflow Visualization
Application Note: Surface Functionalization of Nanocarriers for Targeted Drug Delivery
In nanomedicine, surface functionalization of nanocarriers (e.g., liposomes, dendrimers, mesoporous silica) is paramount for achieving active targeting to tumor sites [50]. This involves decorating the nanoparticle surface with targeting ligands that recognize and bind to receptors overexpressed on cancer cells. This strategy enhances the specificity of therapeutic agents, improves cellular uptake, and minimizes off-target effects, thereby addressing key limitations in conventional cancer therapy [50].
Detailed Experimental Protocol: Functionalization of Liposomes with Targeting Ligands
- Objective: To conjugate a thiol-containing targeting ligand (e.g., an antibody fragment or peptide) to maleimide (MAL)-functionalized liposomes.
- Materials: Pre-formed liposomes containing MAL-PEG-lipid conjugates, Targeting ligand with a free thiol (-SH) group, Phosphate Buffered Saline (PBS), Purification equipment (e.g., dialysis membrane, size exclusion chromatography).
- Procedure:
- Prepare Reactive Liposomes: Use pre-formed liposomes that incorporate MAL-PEG-lipid conjugates, presenting maleimide groups on their surface.
- Ligand Conjugation:
- Dissolve or dialyze the thiol-containing targeting ligand into a suitable buffer (e.g., degassed PBS, pH ~7.0-7.4). Avoid using thiol-containing buffers.
- Add the ligand solution to the liposome suspension under gentle stirring. A typical molar excess of ligand to maleimide groups may be used.
- Allow the reaction to proceed for several hours (e.g., 4-6 h) at room temperature or overnight at 4°C.
- Purification: Remove unreacted ligands by dialyzing the liposome suspension against a large volume of buffer or by using size exclusion chromatography.
- Verification: Confirm successful conjugation using techniques such as gel electrophoresis, HPLC, or spectrophotometric assays [50].
Results and Data Analysis
Functionalized nanocarriers demonstrate significantly improved targeting efficacy. For instance, transferrin-conjugated nanodiamonds showed successful internalization into HeLa cells via receptor-mediated endocytosis, enabling target-specific delivery [53]. The table below summarizes common ligand types and their applications.
Table 3: Targeting Ligands for Nanocarrier Functionalization
| Ligand Type | Example | Target / Receptor | Application in Cancer Therapy |
|---|---|---|---|
| Antibodies / Fragments | Anti-EGFR fragments | Epidermal Growth Factor Receptor (EGFR) | Targeted delivery to various epithelial cancers [50] |
| Peptides | RGD peptide | αvβ3 Integrin | Targeting tumor vasculature and metastatic cells [50] |
| Vitamins | Folic Acid | Folate Receptor | Targeting overexpressed folate receptors on many cancer cells [50] |
| Aptamers | DNA/RNA aptamers | Specific membrane proteins | High-affinity targeting of various cancer cell types [50] |
| Proteins | Transferrin | Transferrin Receptor | Exploiting the high iron demand of cancer cells [53] |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Surface Functionalization Protocols
| Reagent / Material | Function / Role | Example Use Case |
|---|---|---|
| 3-Aminopropyltriethoxysilane (APTES) | Silane coupling agent; introduces primary amine groups to inorganic surfaces. | First-step functionalization of ZrO₂, SiO₂ nanoparticles [48]. |
| N,N'-Diisopropylcarbodiimide (DIC) | Carbodiimide coupling agent; activates carboxyl groups for amide bond formation. | Coupling carboxylic acids to amine-functionalized ZrO₂ nanoparticles [48]. |
| Maleimide (MAL)-PEG-Lipid | Amphiphilic conjugate; incorporates maleimide groups on liposome surface for thiol coupling. | Functionalization of liposomes for conjugation with thiolated antibodies/peptides [50]. |
| EDC / NHS | Zero-length crosslinkers; activate carboxyl groups for efficient amide bond formation with amines. | Conjugation of biomolecules to hydrolyzed polymer scaffolds (e.g., PLGA) [49]. |
| 1,6-Hexanediamine | Alkyl diamine; used in aminolysis to introduce primary amine groups onto polyester surfaces. | Surface functionalization of PCL and PLLA nanofibers [49]. |
| Sodium Hydroxide (NaOH) | Strong base; used in hydrolysis to cleave ester bonds and generate carboxyl groups. | Surface activation of PLGA, PET, and PLA polymers [49]. |
Workflow Visualization
Polymeric nano-biocomposites (PNBs) represent a transformative class of materials derived from the combination of polymer-polymer or nano-scale fillers and polymers, where fillers may include organic/inorganic clays, metal nanoparticles, and hydroxyapatite [54]. These materials constitute a fascinating multidisciplinary area bridging material science, biological science, and nanotechnology with significant impact on medical science [54]. The global shift toward a circular economy has intensified demand for smart polymeric materials that are not only functional but also sustainable, biodegradable, and biocompatible [55].
Controlled drug delivery systems utilizing polymer nanocomposites enable precise pharmaceutical ingredient release to achieve desired therapeutic responses while overcoming limitations of conventional drug delivery systems that suffer from poor bioavailability, plasma drug level fluctuations, and inability to achieve sustained release [56]. By circumventing physiological barriers such as the gastrointestinal tract and the blood-brain barrier, these advanced systems enhance bioavailability and therapeutic efficacy while reducing systemic side effects [57]. The integration of stimuli-responsive capabilities further allows for controlled, often reversible variations in chemical structures or functions in response to external triggers such as pH, temperature, magnetic fields, and mechanical forces [55].
Table 1: Classification of Nano-Biocomposite Structures Based on Dimensionality
| Dimensional Classification | Structural Description | Key Properties and Applications |
|---|---|---|
| 0D (Zero-dimensional) | Spherical nano-clusters or nano-dispersion | Nano-scale in all dimensions; used for diagnostic applications and uniform drug encapsulation |
| 1D (One-dimensional) | Tube-shaped structures (nanometers thick, 100-1000 nm long) | High aspect ratios effective for transporting charged ions and active drugs along controlled directions |
| 2D (Two-dimensional) | Nano-sheets with two dimensions at nanoscale | Large surface area for enhanced biological membrane interaction; layered structures for controlled release |
| 3D (Three-dimensional) | Iso-dimensional nano-biocomposites forming polycrystalline systems | Complex architectures for multi-stage drug release; scaffold structures for tissue engineering |
Engineering Principles of Stimuli-Responsive Nanocomposites
Fundamental Release Mechanisms
Stimuli-responsive polymers (SRPs) undergo controlled variations in their chemical structures or functions in response to single or multiple external stimuli, making them ideal for targeted drug delivery, environmental remediation, soft actuators, and adaptive devices [55]. These intelligent materials can be designed to respond to specific physiological conditions or external triggers, enabling precise spatial and temporal control over drug release.
The design of effective stimuli-responsive nanocomposites requires careful consideration of material properties, trigger mechanisms, and release kinetics. pH-responsive systems exploit variations in physiological pH environments (such as the acidic tumor microenvironment or gastrointestinal tract) to trigger drug release. Temperature-responsive polymers undergo conformational changes or phase transitions at specific temperature thresholds, while magnetic-responsive composites can be activated through external magnetic fields for targeted delivery and controlled release [55].
Nanocomposite Structural Design
Polymer nanocomposites can be categorized into three main groups based on matrix material: polymer matrix nanocomposites (PMNC), metal matrix nanocomposites (MMNC), and ceramic matrix nanocomposites (CMNC) [58]. For drug delivery applications, polymeric and metal nanocomposites are preferred as nanocarriers [58].
Based on interfacial interactions and structural organization, nano-biocomposites are further classified into:
- Intercalated nanocomposites: Feature extended polymer intercalated between host polymers in well-ordered multilayers, with properties similar to ceramic materials [54].
- Exfoliated nanocomposites: Contain extended polymer layers separated in a continuous polymer matrix with mean space dependent on filler loading, typically requiring lower filler amounts than other types [54].
- Flocculated nanocomposites: Similar to intercalated composites but with different arrangement of extended polymer within host polymer [54].
Advanced Fabrication Methods for Genuine Nanocomposites
Innovative "Creating Instead of Adding" Approach
Conventional manufacturing of polymer nanocomposites through simple blending of matrix and reinforcement often fails to achieve predicted mechanical properties due to insufficient dispersion of nano-sized materials [24]. To overcome the fundamental thermodynamic hurdles in achieving good dispersion, innovative techniques that bypass the dispersion step entirely have been developed.
The "creating instead of adding" method represents a paradigm shift in nanocomposite manufacturing [24]. This approach entails commencing the manufacturing process with one component while producing the second component concurrently. The cold drawing process of a polymer blend transforms the minor component into uniformly dispersed nanofibrils, yielding a nanofibrillar polymer-polymer composite as the final material. Selective extraction of the matrix component from the cold drawn blend leads to the formation of neat nanofibrils, which post-compression molding are converted into nanofibrillar single polymer composites [24]. This methodology can potentially boost tensile strength and modulus by 300-400%, or even up to ten times more than older techniques, by bypassing the dispersion step and ensuring optimal distribution of nano-sized reinforcement throughout the polymer [24].
Established Fabrication Techniques
Various sophisticated preparation methods have been developed for creating nanocomposites with enhanced performance characteristics [58]:
- In situ polymerization: The nanofiller is dispersed in the monomer followed by polymerization, enabling excellent filler distribution and strong matrix-filler interactions.
- Intercalation techniques: Particularly for layered fillers, where polymer chains penetrate between filler layers to create ordered structures.
- Sol-gel methods: Involve the transition from a liquid solution to a solid network through hydrolysis and condensation reactions, ideal for ceramic-based nanocomposites.
- Hydrothermal synthesis: Uses high-temperature water under pressure to crystallize materials directly from solution, effective for creating well-defined nanostructures.
- Melt blending and solution blending: Traditional but effective methods where polymers are directly mixed with nanofillers either in molten state or in solution [54].
Table 2: Advanced Fabrication Techniques for Polymer Nanocomposites
| Fabrication Method | Key Mechanism | Advantages | Limitations |
|---|---|---|---|
| Cold Drawing with In-situ Fibrillation | Transforms minor blend component into nanofibrils during drawing | Bypasses dispersion challenges; enhances mechanical properties 300-400% | Limited to specific polymer combinations; requires precise processing control |
| In-situ Polymerization | Polymerization around pre-dispersed nanofillers | Excellent filler distribution; strong matrix-filler bonding | Potential for uncontrolled polymerization; residual monomer removal |
| Intercalation Techniques | Polymer chains penetrate between filler layers | Ordered structures with enhanced properties | Limited to layered fillers; may require compatibilizers |
| Sol-Gel Process | Transition from solution to solid network through hydrolysis | High purity; homogeneous distribution; low processing temperature | Shrinkage issues; residual solvents; limited thickness |
| Selective Laser Sintering | Additive manufacturing using laser to fuse powder materials | Complex geometries; customized implants; no need for supports | High equipment cost; limited material selection; surface roughness |
Automation and Data-Driven Design
Recent advances in automation and data-driven design have revolutionized polymer therapeutics development. High-throughput experimentation (HTE) and screening (HTS) approaches enable rapid exploration of large chemical spaces to identify critical structure-property relationships [59]. With modern controlled living radical polymerization (CLRP) techniques, polymers can be precisely tuned through parameters including degree of polymerization, composition, architecture, stereochemistry, compactness, and valency [59].
Artificial intelligence (AI) and machine learning (ML) applications are emerging to address limitations of traditional HTE and HTS, providing experimental feedback mechanisms to incrementally modify study elements and efficiently sample diverse combinatorial libraries [59]. These data-driven approaches facilitate the design of polymers with specific characteristics optimized for gene delivery, drug delivery, antimicrobial polymer therapeutics, and bioactive polymers including polymer-peptide, polymer-nucleic acid, polymer-drug, and protein-polymer conjugates [59].
Experimental Protocols
Protocol 1: Fabrication of pH-Responsive Polymeric Nanocomposite Films for Targeted Drug Delivery
Objective: Prepare and characterize alginate-chitosan nanocomposite films loaded with model drug (e.g., doxorubicin) for pH-responsive release in tumor microenvironments.
Materials:
- Sodium alginate (low viscosity)
- Chitosan (medium molecular weight)
- Citric acid (crosslinking agent)
- Model drug (doxycycline hydrochloride or similar)
- Acetic acid (1% v/v solution)
- Calcium chloride (2% w/v solution)
- Deionized water
Equipment:
- Magnetic stirrer with hotplate
- Ultrasonic bath sonicator
- Centrifuge
- Vacuum oven
- UV-Vis spectrophotometer
- FTIR spectrometer
- Scanning Electron Microscope
Procedure:
Polymer Solution Preparation:
- Dissolve 2g sodium alginate in 100mL deionized water with continuous stirring at 500rpm, 60°C for 4h
- Separately, dissolve 1g chitosan in 100mL 1% acetic acid solution with stirring at 400rpm, 40°C for 6h
Nanocomposite Formation:
- Mix alginate and chitosan solutions in 3:1 volume ratio
- Add 0.5g citric acid as crosslinking agent
- Incorporate drug payload (5-10% w/w of polymer content)
- Sonicate mixture at 40kHz, 60°C for 30min to ensure homogeneous dispersion
Film Casting and Crosslinking:
- Pour 20mL of nanocomposite solution into Petri dishes (9cm diameter)
- Dry at 40°C in vacuum oven for 12h
- Immerse formed films in 2% CaCl₂ solution for 30min to enhance crosslinking
- Rinse with deionized water and dry at room temperature for 24h
Characterization:
- Analyze chemical structure using FTIR (4000-400cm⁻¹ range)
- Examine surface morphology by SEM at 10kV acceleration voltage
- Determine drug loading efficiency by UV-Vis spectrophotometry
Quality Control Parameters:
- Film thickness: 0.1±0.02mm
- Drug loading efficiency: >85%
- Uniformity of drug distribution: RSD <5%
Protocol 2: Evaluation of Stimuli-Responsive Drug Release Profiles
Objective: Quantify drug release kinetics from nanocomposite films under varying pH conditions simulating physiological environments.
Materials:
- Prepared drug-loaded nanocomposite films
- Phosphate buffered saline (PBS) pH 7.4
- Acetate buffered saline (ABS) pH 5.0
- Model drug for standard curve
- Dialysis membranes (MWCO 12-14kDa)
Equipment:
- UV-Vis spectrophotometer
- Dissolution apparatus with paddles
- Thermostatic water bath
- HPLC system (optional for verification)
Procedure:
Standard Curve Preparation:
- Prepare stock solution of model drug (1mg/mL)
- Create serial dilutions (0, 2, 5, 10, 20, 50μg/mL)
- Measure absorbance at λmax (drug-dependent)
- Plot concentration vs. absorbance with R²>0.99
Drug Release Study:
- Cut films into 1×1cm squares
- Place in dialysis bags containing 5mL release medium
- Immerse in 200mL PBS pH 7.4 or ABS pH 5.0 at 37±0.5°C
- Agitate at 50rpm in dissolution apparatus
- Withdraw 2mL samples at predetermined intervals (0.5, 1, 2, 4, 6, 8, 12, 24h)
- Replace with fresh medium to maintain sink conditions
Analysis:
- Measure sample absorbance using UV-Vis spectrophotometer
- Calculate cumulative drug release using standard curve
- Plot release profiles for different pH conditions
Release Kinetics Modeling:
- Fit data to zero-order, first-order, Higuchi, and Korsmeyer-Peppas models
- Determine release mechanism based on model parameters
Data Interpretation:
- pH-sensitive systems should show significantly faster release at pH 5.0 versus pH 7.4
- Higuchi model indicates diffusion-controlled release
- Korsmeyer-Peppas exponent (n) values:
- n < 0.45: Fickian diffusion
- 0.45 < n < 0.89: Anomalous transport
- n > 0.89: Case-II transport
Table 3: Characterization Techniques for Nanocomposite Drug Delivery Systems
| Characterization Technique | Information Obtained | Experimental Conditions | Data Interpretation |
|---|---|---|---|
| X-ray Diffraction (XRD) | Crystallinity, phase identification, intercalation | 2θ range: 5-80°; Cu Kα radiation | Shift in peaks indicates polymer-filler interaction; broadening suggests nanoscale dimensions |
| Scanning Electron Microscopy (SEM) | Surface morphology, filler distribution, defects | Acceleration voltage: 5-20kV; sputter coating with gold | Homogeneous distribution indicates good dispersion; agglomerates suggest poor compatibility |
| Transmission Electron Microscopy (TEM) | Internal structure, filler dispersion, interface | Acceleration voltage: 80-200kV; ultrathin sections | Direct visualization of nanofiller arrangement; intercalated or exfoliated structures |
| Fourier-Transform Infrared Spectroscopy (FTIR) | Chemical structure, functional groups, interactions | Range: 4000-400cm⁻¹; resolution: 4cm⁻¹ | Peak shifts indicate hydrogen bonding or chemical interactions between components |
| Thermogravimetric Analysis (TGA) | Thermal stability, decomposition profile | Temperature range: 25-800°C; heating rate: 10°C/min | Increased decomposition temperature indicates enhanced thermal stability |
| Differential Scanning Calorimetry (DSC) | Thermal transitions, glass transition, crystallinity | Temperature range: -50 to 300°C; heating rate: 10°C/min | Changes in Tg indicate polymer-filler interactions; melting peaks show crystallinity changes |
Research Reagent Solutions Toolkit
Table 4: Essential Research Reagents for Nanocomposite Drug Delivery Systems
| Reagent Category | Specific Examples | Function in Formulation | Application Notes |
|---|---|---|---|
| Natural Polymers | Alginate, Chitosan, Starch | Biocompatible matrix; mucoadhesive properties; biodegradability | Alginate popular for grafting, blending, derivatization; Chitosan cost-effective and environmentally friendly [54] |
| Synthetic Polymers | PLA, PGA, PLGA, Polyanhydrides | Controlled degradation rates; tunable mechanical properties | PLGA most common synthetic polymer for controlled delivery; degradation rate adjustable via lactide:glycolide ratio |
| Stimuli-Responsive Polymers | pH-sensitive: Poly(acrylic acid); Temperature-sensitive: PNIPAM | Environment-responsive drug release; targeted delivery | PNIPAM exhibits LCST around 32°C; expands below and collapses above this temperature |
| Nanofillers | Montmorillonite clay, Hydroxyapatite, Metal nanoparticles | Enhanced mechanical strength; modified release profiles; additional functionality | Clay improves hydrogel water retention; metal nanoparticles enable magnetic/thermal responsiveness [54] [58] |
| Crosslinking Agents | Glutaraldehyde, Citric acid, Genipin | Improve structural integrity; control swelling and degradation | Citric acid offers low toxicity; genipin natural alternative with blue pigment formation |
| Solvents | Ionic liquids, Water, Ethanol | Green processing; maintain protein stability | Ionic liquids as green solvents for renewable/bio-derived feedstocks [55] |
Implementation and Validation Workflows
Integration with Advanced Manufacturing Technologies
Additive manufacturing (3D printing) technologies enable fabrication of complex, patient-specific drug delivery devices with precise spatial control over composition and architecture [55]. Photopolymerization-based approaches using digital light processing have been successfully employed to create high-performance zirconia ceramic components with excellent mechanical properties [7]. These technologies allow for customized dosage forms with complex release profiles tailored to individual patient needs.
The development of fully recyclable composites has spurred extensive research into thermoplastics and their blends, valued for their recyclability and excellent mechanical properties [7]. Recent investigations into stress-strain behavior of polymer blends such as PES/PEEK, PPS/PEEK, and HDPE/PP have provided valuable insights into the mechanical viability and sustainability benefits of using recycled thermoplastic blends as matrices for unidirectional fiber-reinforced composites [7].
Validation and Translation Considerations
For successful clinical translation, nanocomposite drug delivery systems must undergo comprehensive evaluation including:
- In vitro release studies under physiological conditions to establish release kinetics
- Cytocompatibility assessment using relevant cell lines to ensure safety
- Hemocompatibility testing for systems intended for intravenous administration
- Stability studies under various storage conditions to determine shelf life
- Sterilization validation to ensure maintenance of structure and function after sterilization
Advanced modeling approaches, including Representative Volume Element-based simulations, enable prediction of mechanical properties of unidirectional fiber-reinforced composites, providing valuable preliminary insight before extensive experimental work [7]. Multi-scale simulations and parametric analyses help optimize performance characteristics for specific applications, such as seismic-resistant building components or radiation protection materials [7].
Active implantable drug delivery systems represent particularly advanced applications, requiring integration of power systems, communication protocols, and biocompatible materials to achieve sustained or on-demand drug release, remote activation, and programmable dosing [57]. These systems enhance patient compliance while minimizing intervention frequency through precise drug administration tailored to individual patient needs [57].
The development of advanced coatings for medical devices and implants represents a critical frontier in the fight against microbial infection and biofilm formation. Polymer nanocomposites, which integrate nanoscale fillers within a polymer matrix, have emerged as transformative materials in this domain [60]. They synergize the structural versatility and biocompatibility of polymers with the potent antimicrobial properties of various nanofillers, offering a robust strategy to combat implant-associated infections (IAIs) [4] [61]. These infections are particularly challenging to treat as biofilms can exhibit resistance to antibiotics at levels 1000 to 1500 times higher than their planktonic counterparts [61]. This application note details the latest advances and provides explicit protocols for developing and evaluating antimicrobial and antibiofilm nanocomposite coatings, framed within a broader research context on polymer nanocomposites fabrication.
Background and Significance
The Challenge of Biofilms and Implant-Associated Infections
Biofilms are structured communities of microorganisms encapsulated within a self-produced matrix of extracellular polymeric substances (EPS) that adhere to biological or abiotic surfaces [61]. An estimated 65–80% of microbial infections and 80% of chronic human infections are linked to biofilm formation [61]. On surgical implants, biofilms can lead to device failure and severe patient outcomes. The biofilm matrix presents a formidable barrier, restricting antibiotic penetration and rendering embedded bacteria highly tolerant to conventional treatments [61] [62]. Furthermore, the biofilm microenvironment facilitates the horizontal transfer of drug-resistant genes, compounding the problem of antimicrobial resistance (AMR) [62].
The Promise of Polymer Nanocomposite Coatings
Polymer nanocomposites offer a multifaceted solution to these challenges. By incorporating nanofillers such as silver nanoparticles (AgNPs), zirconia (ZrO₂), graphene oxide (GO), or other functional nanomaterials, these coatings can be engineered to possess:
- Inherent antimicrobial activity through the release of ions or generation of reactive oxygen species (ROS) [60] [62].
- Enhanced physical barriers against microbial adhesion and corrosion [63].
- Stimuli-responsive functionality for on-demand antimicrobial activity, for example, via photothermal or photodynamic therapy [61] [64]. The polymer matrix serves to stabilize the nanoparticles, control the release kinetics of antimicrobial agents, and improve the overall biocompatibility of the coating [60].
Key Nanocomposite Coating Systems and Quantitative Performance
Recent research has yielded several promising coating systems with demonstrated efficacy against biofilms. The table below summarizes the composition and performance of key nanocomposite coatings from recent studies.
Table 1: Performance Summary of Recent Antimicrobial Nanocomposite Coatings
| Coating System | Substrate | Key Components | Antimicrobial Mechanism | Reported Efficacy | Reference |
|---|---|---|---|---|---|
| HAp-ZrO₂-GO Nanocomposite | 316L Stainless Steel | Hydroxyapatite, Zirconia, Graphene Oxide | Enhanced corrosion resistance, antibacterial activity, bioactivity | Significant increase in corrosion resistance; potent antibacterial action; confirmed biocompatibility. | [63] |
| AgBiS₂@CQDs/Ti | Medical Titanium | Silver Bismuth Sulfide, Carbon Quantum Dots | Photodynamic Therapy (PDT) & Photothermal Therapy (PTT) under NIR-II laser | Eradication of bacteria and rapid destruction of mature biofilm under 1064 nm NIR laser irradiation. | [64] |
| AgNP-Polymer Nanocomposites (AgNP-PNCs) | Various Polymer Matrices | Silver Nanoparticles, Polymer Matrix | Controlled release of Ag⁺ ions; generation of ROS; membrane disruption. | Enhanced infection control for wound dressings, medical coatings, and tissue scaffolds. | [60] |
Detailed Experimental Protocols
Protocol: Synthesis of a HAp-ZrO₂-GO Nanocomposite Coating on 316L SS
This protocol is adapted from a study developing a coating for hammertoe implants [63].
Research Reagent Solutions
Table 2: Essential Materials for HAp-ZrO₂-GO Coating Fabrication
| Reagent/Material | Specification/Purity | Function in the Protocol |
|---|---|---|
| 316L Stainless Steel (SS) | Medical grade, cut to desired implant dimensions | Substrate for the coating. |
| Graphene Oxide (GO) | Aqueous dispersion, high concentration | Enhances corrosion resistance and provides a platform for composite formation. |
| Zirconia (ZrO₂) Nanopowder | High-purity, nanoscale | Improves mechanical strength and toughness of the coating. |
| Hydroxyapatite (HAp) Powder | Synthetic, nanoscale | Provides bioactivity and improves biocompatibility. |
| Ethanol & Acetone | Analytical Reagent (AR) Grade | For cleaning and degreasing the substrate surface. |
| Simulated Body Fluid (SBF) | Prepared as per Kokubo recipe | For in vitro corrosion resistance and bioactivity testing. |
Step-by-Step Methodology
Substrate Preparation:
- Cut the 316L SS into coupons (e.g., 10 mm x 10 mm x 1 mm).
- Mechanically polish the coupons with successive grades of silicon carbide paper up to 2000 grit to achieve a uniform surface finish.
- Clean the polished coupons by sonication in acetone for 15 minutes, followed by ethanol for 15 minutes to remove organic contaminants.
- Dry the substrates in a clean oven at 60°C for 1 hour.
Suspension Preparation:
- Prepare a homogeneous suspension containing 1-2 wt% GO in a mixture of deionized water and ethanol.
- Add nanoscale ZrO₂ powder and nanoscale HAp powder to the GO suspension. The typical ratio can be optimized around 1:1:1 (HAp:ZrO₂:GO by weight).
- Use probe sonication for 30-60 minutes to ensure uniform dispersion of the nanomaterials and prevent agglomeration.
Coating Deposition:
- Employ a spin-coating or dip-coating method to apply the nanocomposite suspension onto the pre-treated 316L SS substrates.
- For spin-coating, deposit a few drops of the suspension and spin at 2000-3000 rpm for 30 seconds.
- Repeat the coating process 3-5 times to build up a uniform coating thickness, with intermediate drying at 80°C for 10 minutes between each layer.
Post-deposition Treatment:
- Sinter the coated substrates in a furnace at a temperature of 800-900°C for 2 hours in an inert argon atmosphere.
- The heating and cooling rates should be controlled (e.g., 5°C/min) to minimize thermal stress and prevent cracking of the coating.
Characterization and Evaluation Workflow
The following diagram outlines the key steps for fabricating and characterizing the nanocomposite coating.
Protocol: Evaluating Antibacterial and Antibiofilm Efficacy
This is a standard protocol for assessing the biological performance of coatings [63] [61].
Materials
- Test strains: Staphylococcus aureus (ATCC 6538) and Escherichia coli (ATCC 8739).
- Culture media: Tryptic Soy Broth (TSB), Nutrient Agar.
- Phosphate Buffered Saline (PBS), pH 7.4.
- Live/Dead BacLight Bacterial Viability Kit (or similar).
- 24-well cell culture plate.
Methodology
Biofilm Cultivation:
- Grow bacterial cultures overnight in TSB to the mid-logarithmic phase.
- Dilute the cultures to a concentration of ~1×10⁶ CFU/mL in fresh TSB.
- Place sterile coated and uncoated (control) substrates into the wells of a 24-well plate.
- Add 2 mL of the bacterial suspension to each well.
- Incubate the plate at 37°C for 24-48 hours under static conditions to allow biofilm formation.
Antibacterial Assay (Post-incubation):
- After incubation, carefully remove each substrate from the well using sterile forceps.
- Gently rinse with PBS to remove non-adherent planktonic cells.
- Transfer the substrate to a tube containing 10 mL of PBS.
- Sonicate the tube for 10-15 minutes to disaggregate and detach the biofilm from the surface.
- Serially dilute the resulting suspension and plate on Nutrient Agar plates.
- Count the colony-forming units (CFUs) after 24 hours of incubation at 37°C.
- Calculate the percentage reduction in bacterial viability compared to the uncoated control.
Biofilm Visualization (Live/Dead Staining):
- After rinsing the biofilm-coated substrates with PBS, stain with the Live/Dead stain mixture (e.g., SYTO 9 and propidium iodide) according to the manufacturer's instructions.
- Incubate in the dark for 15-20 minutes.
- Visualize using a confocal laser scanning microscope (CLSM). Live bacteria will stain green, and dead/damaged bacteria will stain red.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Key Reagents for Nanocomposite Coating Development and Evaluation
| Category / Item | Specific Examples | Primary Function in R&D |
|---|---|---|
| Nanomaterials | ||
| Silver Nanoparticles (AgNPs) | AgNP powder, Ag⁺ precursors | Potent broad-spectrum antimicrobial agent; disrupts cell membranes and generates ROS. |
| Graphene Oxide (GO) | GO sheets, GO dispersions | Provides a high-surface-area platform; enhances mechanical properties and corrosion resistance. |
| Zirconia (ZrO₂) | ZrO₂ nanopowder | Improves coating toughness, wear resistance, and biocompatibility. |
| Carbon Quantum Dots (CQDs) | CQDs suspension | Acts as a charge-transfer bridge in photocatalytic systems; enhances photodynamic therapy. |
| Polymer Matrices | ||
| Biopolymers | Chitosan, Alginate, Polyvinyl Alcohol (PVA) | Provides biodegradable, biocompatible matrix for controlled drug release and coating formation. |
| Characterization Tools | ||
| Surface Analysis | SEM, EDX, FTIR | Analyzes coating morphology, elemental composition, and chemical bonding. |
| Biological Assays | Live/Dead Staining, CFU Count | Quantifies antibacterial efficacy and biofilm viability on coated surfaces. |
| Corrosion Testing | Electrochemical Workstation, SBF | Evaluates coating stability and degradation in physiological environments. |
Advanced Strategies and Future Directions
Stimuli-Responsive and Phototherapeutic Coatings
Beyond passive release systems, advanced coatings can be activated by external stimuli. The AgBiS₂@CQDs/Ti system is a prime example that utilizes a combination of photodynamic therapy (PDT) and photothermal therapy (PTT) [64]. Under NIR-II laser irradiation (1064 nm), the coating generates lethal levels of reactive oxygen species (ROS) and localized high temperatures, synergistically eradicating bacteria and destroying mature biofilms with high spatiotemporal control [64]. The mechanism of this combined action is illustrated below.
Challenges and Clinical Translation
Despite the promising results, several challenges remain for the widespread clinical adoption of these coatings. Key issues include:
- Long-term cytotoxicity and biocompatibility of leached nanoparticles, which are dependent on particle size, shape, and ion release kinetics [60].
- Scalability and reproducibility of synthesis and coating techniques for industrial-level production [60] [24].
- Complex regulatory pathways due to uncertainties regarding long-term toxicity, environmental impact, and potential bacterial resistance to nanomaterials [60].
Future research should focus on optimizing hybrid strategies, developing "smart" coatings with feedback-controlled antimicrobial release, and conducting extensive preclinical and clinical studies to ensure safety and efficacy [60] [65].
The field of tissue engineering has emerged as a revolutionary approach for regenerating damaged tissues and organs, offering solutions to challenges posed by traditional transplants. Central to this paradigm are scaffolds, three-dimensional structures that serve as temporary templates to support cell attachment, proliferation, and differentiation [66]. The integration of nanotechnology has been pivotal in advancing scaffold design, leading to the development of polymer nanocomposites [4]. These materials combine a biodegradable polymer matrix with nanoscale fillers, resulting in scaffolds with superior mechanical properties, enhanced bioactivity, and improved biocompatibility compared to conventional materials [4] [66]. This application note details the fabrication, characterization, and evaluation of advanced polymeric nanocomposite scaffolds, providing a standardized protocol for researchers in academia and industry. The content is framed within a broader research thesis on polymer nanocomposites fabrication methods, emphasizing practical methodology and data analysis for drug development professionals and tissue engineering scientists.
Key Nanocomposite Systems for Scaffold Fabrication
Polymer nanocomposites are classified based on the nature of the nanofiller incorporated into the polymer matrix. The choice of system dictates the final scaffold's physicochemical and biological properties.
- Organic Nanocomposites: These incorporate carbon-based nanomaterials such as carbon nanotubes (CNTs) and graphene oxide (GO). CNTs contribute exceptional mechanical strength and electrical conductivity, which is beneficial for neural and cardiac tissue engineering [67]. GO, with its high surface area and functional groups, improves mechanical strength and offers sites for further biofunctionalization [68].
- Inorganic Nanocomposites: This category includes ceramic nanoparticles like hydroxyapatite (HAp) and metal oxides such as cerium oxide ( nanoceria). HAp, a natural component of bone, enhances osteoconductivity and mechanical strength in bone graft substitutes [69] [68]. Nanoceria acts as an antioxidant, scavenging free radicals to mitigate oxidative stress and support cell survival in inflamed or damaged tissues [69].
- Bio-based Green Nanocomposites: Developed from sustainable resources like chitosan, alginate, and cellulose, these composites are prized for their biocompatibility, biodegradability, and low immunogenicity. They are often combined with inorganic nanofillers to create "green" scaffolds with enhanced functionality [4].
Table 1: Key Nanocomposite Systems and Their Properties in Tissue Engineering
| Nanocomposite System | Key Components | Enhanced Scaffold Properties | Primary Tissue Applications |
|---|---|---|---|
| CNT-Polymer [67] | MWCNT/SWCNT, Chitosan, Poly(lactic-co-glycolic acid) | Electrical conductivity, Tensile strength, Drug loading capacity | Neural, Cardiac, Bone |
| HAp-Polymer [69] [68] | Hydroxyapatite, Alginate, Gelatin, PVA | Compressive strength, Osteoconductivity, Biomineralization | Bone, Dental |
| GO-Polymer [68] | Graphene Oxide, Clay, PVA, Fe₃O₄ | Mechanical strength, Antibacterial activity, Osteoinductivity | Bone, Skin |
| Bio-based Green [4] | Chitosan, Gelatin, Alginate, Cellulose | Biocompatibility, Biodegradability, Low immunogenicity | Cartilage, Skin, Bone |
Experimental Protocols
Synthesis of a PVA/CMC/HAp/CGF Nanocomposite Scaffold
This protocol describes the fabrication of a multifunctional scaffold using a natural-synthetic polymer blend reinforced with hydroxyapatite and magnetic clay-graphene oxide for bone tissue engineering, adapted from recent research [68].
3.1.1 Materials and Reagents
- Polymers: Polyvinyl Alcohol (PVA), Carboxymethyl Cellulose (CMC) sodium salt.
- Nanofillers: Natural Hydroxyapatite (HAp) powder, Magnetic Clay modified with Graphene Oxide (CGF). Synthesis of CGF involves co-precipitation of Fe₃O₄ with clay and GO using the modified Hummer's method [68].
- Solvent: Deionized water.
- Equipment: Magnetic stirrer, Sonicator, Freeze-dryer, Mold.
3.1.2 Step-by-Step Procedure
- Solution Preparation: Dissolve PVA (e.g., 2% w/v) and CMC (e.g., 1% w/v) separately in deionized water under constant stirring at 80°C for 4 hours until clear solutions are obtained.
- Polymer Blending: Combine the PVA and CMC solutions in a desired mass ratio (e.g., 70:30) and stir for another 2 hours at 60°C to form a homogeneous polymer blend.
- Nanofiller Dispersion:
- Weigh out 10 wt.% HAp and 2 wt.% CGF relative to the total polymer weight.
- Disperse the nanofillers in a small volume of deionized water using probe sonication (e.g., 400 W, 30 minutes, pulse mode) to achieve a homogeneous suspension.
- Nanocomposite Mixing: Gradually add the nanofiller suspension to the polymer blend under vigorous mechanical stirring, followed by further sonication for 15 minutes to ensure uniform dispersion and avoid agglomeration.
- Casting and Cross-linking: Pour the final mixture into a polytetrafluoroethylene (PTFE) mold. The scaffold can be ionically cross-linked by exposing it to a divalent cation solution (e.g., CaCl₂) if alginate is used, or physically stabilized.
- Freeze-Drying: Rapidly freeze the cast scaffold at -80°C for 6 hours, then transfer to a freeze-dryer for 48 hours to sublime the ice crystals, creating a porous 3D structure.
- Storage: Store the dried scaffolds in a desiccator at room temperature until further use.
Scaffold Characterization and Biocompatibility Assessment
Rigorous characterization is essential to confirm the scaffold meets the desired physicochemical and biological criteria.
3.2.1 Physicochemical Characterization
- Porosity Analysis: Use liquid displacement method with ethanol. Porosity (%) = [(V1 - V3) / (V2 - V3)] * 100, where V1 is the volume of ethanol after scaffold immersion, V2 is the initial ethanol volume, and V3 is the volume after scaffold removal. Target porosity for bone scaffolds is >70% [68].
- Swelling Ratio: Weigh dry scaffold (Wd), immerse in Phosphate Buffered Saline (PBS, pH 7.4) at 37°C, and weigh at regular intervals after removing surface moisture (Ws). Swelling Ratio = (Ws - Wd) / Wd.
- Compressive Strength: Perform uniaxial compression test on cylindrical scaffold samples using a universal testing machine at a fixed crosshead speed. Record the maximum stress at failure.
- Biomineralization: Incubate scaffold in Simulated Body Fluid (SBF) at 37°C for 14 days. Analyze surface for hydroxyapatite crystal formation using Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDS) [68].
3.2.2 In Vitro Biocompatibility Assay (MTT Assay)
- Sterilization: Sterilize scaffolds under UV light for 1 hour per side.
- Cell Seeding: Seed pre-osteoblast cells (e.g., MC3T3-E1) onto scaffolds at a density of 1x10⁴ cells/scaffold in 24-well plates.
- Incubation: Culture cells in standard conditions (37°C, 5% CO₂) for 1, 3, and 7 days.
- MTT Incubation: At each time point, add MTT reagent (0.5 mg/mL) to each well and incubate for 4 hours.
- Solubilization: Carefully remove the medium and add dimethyl sulfoxide to dissolve the formed formazan crystals.
- Absorbance Measurement: Transfer the solution to a 96-well plate and measure the absorbance at 570 nm using a microplate reader. Cell viability is expressed as a percentage relative to the control group (tissue culture plastic).
Table 2: Typical Experimental Data for PVA-based Nanocomposite Scaffolds [68]
| Scaffold Formulation | Compressive Strength (MPa) | Porosity (%) | Swelling Ratio (%) | Biodegradation (21 days, % mass loss) | Cell Viability (OD at 570nm) |
|---|---|---|---|---|---|
| PVA/CMC/HAp/CGF | 12.0 | 72 | 1860 | 43 | 1.483 |
| PVA/Alginate/HAp/CGF | 8.1 | 79 | - | - | 1.451 |
| Target for Cancellous Bone [68] | 2 - 20 | >70 | N/A | Tunable | > Control |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Nanocomposite Scaffold Fabrication and Testing
| Reagent/Material | Function/Application | Key Characteristics |
|---|---|---|
| Polyvinyl Alcohol (PVA) [68] | Synthetic polymer matrix; provides mechanical strength and stability. | Biocompatible, hydrophilic, forms hydrogels, high chemical stability. |
| Carboxymethyl Cellulose (CMC) [68] | Natural polymer matrix; enhances porosity, swelling, and biodegradability. | Anionic, biocompatible, biodegradable, forms ionic cross-links. |
| Alginate [69] [68] | Natural polymer matrix; forms hydrogels under mild conditions. | Biocompatible, low toxicity, gelling with divalent cations (e.g., Ca²⁺). |
| Hydroxyapatite (HAp) [69] [68] | Bioactive ceramic nanofiller; mimics bone mineral, promotes osteogenesis. | Osteoconductive, improves compressive strength, integrates with bone. |
| Graphene Oxide (GO) [68] | 2D carbon nanofiller; reinforces mechanical structure and adds functionality. | High surface area, antibacterial, improves mechanical properties. |
| Carbon Nanotubes (CNTs) [67] | 1D carbon nanofiller; adds electrical conductivity and mechanical resilience. | High aspect ratio, electrically conductive, requires functionalization for dispersion. |
| Cerium Oxide (Nanoceria) [69] | Antioxidant enzyme-mimic; protects cells from oxidative stress. | Free radical scavenger, cytoprotective, supports cell adhesion and survival. |
| Fe₃O₄ (Magnetite) [68] | Magnetic nanoparticle; enables magnetic targeting and hyperthermia. | Superparamagnetic, biocompatible, used in drug delivery and cell isolation. |
| MTT Reagent [68] | (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide); assesses cell metabolic activity. | Yellow tetrazole reduced to purple formazan in living cells. |
Workflow and Signaling Pathways
Nanocomposite Scaffold Fabrication and Evaluation Workflow
The following diagram outlines the comprehensive process from material preparation to final in vitro assessment, integrating the key protocols described in this document.
Signaling Pathway in Mesenchymal Stem Cell-Mediated Bone Regeneration
This diagram illustrates the key cellular and molecular interactions when a CNT-based nanocomposite scaffold is used to direct the activity of Mesenchymal Stem Cells (MSCs) for bone repair, a critical pathway in regenerative medicine [69].
Overcoming Fabrication Hurdles: Dispersion, Toxicity, and Scalability Challenges
The incorporation of nanofillers into polymer matrices has revolutionized material science, enabling the creation of composites with enhanced mechanical, thermal, electrical, and barrier properties. However, a significant challenge persists: the inherent tendency of nanoparticles to agglomerate. This agglomeration arises from their high surface area-to-volume ratio and strong interparticle forces, such as van der Waals forces and Coulombic interactions, which drive them to form clusters to minimize surface energy [70] [71]. These agglomerates act as defect sites, severely compromising the mechanical integrity and functional performance of the final nanocomposite by creating stress concentration points and hindering efficient load transfer [72]. Consequently, achieving a uniform, molecular-level dispersion of nanofillers is arguably the most critical step in fabricating high-performance polymer nanocomposites. This document outlines the underlying mechanisms of agglomeration and provides detailed, practical protocols for overcoming this pervasive dilemma.
Understanding Agglomeration
Nanoparticle agglomeration is typically categorized into two types, each with distinct characteristics and dispersion challenges.
- Soft Agglomeration: This is primarily caused by weak physical interactions, such as van der Waals forces and electrostatic attraction. These agglomerates are generally reversible and can be disrupted through mechanical means like high-shear mixing or ultrasonication [70] [71].
- Hard Agglomeration: This involves stronger bonds, including chemical bonds and hydrogen bridging, often facilitated by hydroxyl groups on nanoparticle surfaces. Hard agglomerates are not easily broken by mechanical forces alone and require chemical interventions or specialized processing techniques to overcome [71].
The table below summarizes the fundamental differences between these agglomeration types.
Table 1: Types of Nanoparticle Agglomeration
| Agglomeration Type | Dominant Forces | Strength | Dispersion Difficulty | Common Solutions |
|---|---|---|---|---|
| Soft Agglomeration | van der Waals, Coulombic | Weak | Low to Moderate | Mechanical stirring, ultrasonication |
| Hard Agglomeration | Chemical bonds, Hydrogen bonding | Strong | High | Surface chemical modification, mechanochemical methods |
Core Strategies and Protocols
A multi-faceted approach is essential for effectively preventing and mitigating nanofiller agglomeration. The following sections detail the primary strategies.
Chemical Surface Modification
This strategy involves altering the surface chemistry of the nanofiller to improve its compatibility with the polymer matrix and introduce repulsive forces between particles.
Protocol 3.1.1: Surface Functionalization of Nanodiamonds with Polyethyleneimine (PEI) for Enhanced Dispersion
This protocol is adapted from a study on fabricating mixed matrix membranes, where PEI acted as both a dispersing agent and a CO₂ carrier [73].
- Objective: To decorate nanodiamond (ND) surfaces with PEI to minimize agglomeration in a polyether block amide (Pebax) matrix and improve CO₂/N₂ selectivity.
- Materials:
- Nanodiamond: (≥97%, particle size < 10 nm)
- Polyethyleneimine (PEI): (Average M~n~ ~1200, Average M~w~ ~1300)
- Solvent: Deionized water
- Equipment: Ultrasonic bath, centrifuge, vacuum oven, magnetic stirrer with hot plate.
- Procedure:
- Pre-treatment: Heat ND powder at 425°C in air for 4 hours (heating rate 10°C/min) to maximize oxygen-containing functional groups on the surface [73].
- Dissolution: Dissolve 2 g of PEI in 15 mL of deionized water with stirring for 30 minutes to form a homogeneous solution.
- Dispersion: Disperse 0.2 g of the pre-treated ND into the PEI solution. Sonicate the mixture for 15 minutes to achieve a uniform dispersion.
- Reaction: Heat the mixture to 70°C while stirring at 250 rpm for 24 hours to allow the PEI to graft onto the ND surface.
- Purification: Centrifuge the mixture and wash the solid product (ND-PEI) with deionized water at least three times to remove any unreacted PEI.
- Drying: Dry the final ND-PEI product in a vacuum oven at 100°C for 24 hours. Store under vacuum until use [73].
- Key Considerations: The low molecular weight PEI effectively wets the ND surface and acts as an interfacial binder. The formation of an adsorption film provides steric hindrance, preventing particle re-agglomeration.
Hybrid Filler Systems
Combining different types of nanofillers can create synergistic effects that suppress agglomeration, which is particularly effective at high filler loadings.
Protocol 3.2.1: Fabrication of Hybrid GO-hBN/PVA Nanocomposites via Mold-Casting
This protocol leverages the synergistic effect of 2D materials to suppress agglomeration at high filler content, mimicking a nacre-like structure [72].
- Objective: To prepare polyvinyl alcohol (PVA) nanocomposites reinforced with a hybrid of graphene oxide (GO) and hexagonal boron nitride (hBN) to achieve high filler loading without agglomeration.
- Materials:
- Nanofillers: Graphene oxide (GO) nanosheets, hexagonal boron nitride (hBN) flakes.
- Polymer Matrix: Polyvinyl alcohol (PVA) powder (MW ~85,000–124,000).
- Solvent: Deionized water.
- Equipment: Sonicator, magnetic stirrer, vacuum drying oven, mold.
- Procedure:
- Filler Dispersion:
- Prepare separate aqueous dispersions of GO and hBN via sonication.
- Mix the GO and hBN dispersions at the desired mass ratio (e.g., 4:1 GO to hBN) and sonicate further to create a homogeneous hybrid dispersion.
- Matrix Integration:
- Dissolve PVA powder in deionized water at 90°C with vigorous stirring to create a 5 wt% solution.
- Slowly add the hybrid GO-hBN dispersion dropwise into the PVA solution under continuous stirring.
- Maintain stirring for 2-4 hours to ensure a homogeneous mixture.
- Casting and Drying:
- Pour the final mixture into a polytetrafluoroethylene (PTFE) mold.
- Allow the solvent to evaporate at room temperature for 24 hours, followed by further drying in a vacuum oven at 60°C for 24 hours to remove residual solvent [72].
- Filler Dispersion:
- Key Considerations: The incorporation of hBN flakes disrupts the restacking and agglomeration of GO sheets. This hybrid approach allows the filler content to reach up to 80 wt% while still maintaining enhanced mechanical properties, a level where single-filler systems typically fail due to severe agglomeration [72].
Physical Processing and Drying Techniques
The methods used during the final stages of nanoparticle synthesis and processing, particularly drying, are critical for preventing hard agglomeration.
Protocol 3.3.1: Organic Solvent Washing and Azeotropic Distillation for Gel-Derived Nanopowders
This protocol addresses the hard agglomeration caused by capillary forces during the drying of wet gels or precipitates [71].
- Objective: To remove water and surface hydroxyl groups from wet nanoparticle gels to prevent the formation of hard agglomerates during drying.
- Materials:
- Wet gel of nanoparticles (e.g., Al₂O₃, ZrO₂, SiO₂, TiO₂).
- Anhydrous organic solvent (e.g., ethanol, n-butanol).
- Equipment: Centrifuge, distillation apparatus, vacuum oven.
- Procedure:
- Organic Washing:
- Centrifuge the wet gel and discard the aqueous supernatant.
- Re-disperse the gel in an excess of anhydrous ethanol. Agitate or stir for several hours.
- Repeat the centrifugation and washing steps multiple times to ensure complete replacement of water with ethanol.
- Azeotropic Distillation (Alternative):
- For systems where n-butanol is used, employ azeotropic distillation. The mixture of water and n-butanol forms an azeotrope with a boiling point lower than that of pure water, facilitating co-evaporation.
- This process maximally removes water trapped within the colloidal structure.
- Drying: Dry the solvent-exchanged powder in a vacuum oven at a moderate temperature [71].
- Organic Washing:
- Key Considerations: Replacing water with a solvent of lower surface tension reduces capillary forces. Furthermore, the functional groups of the organic solvent (e.g., -OH in ethanol) can passivate the nanoparticle surface by replacing some non-bridging hydroxyl groups, providing steric hindrance and reducing the opportunity for strong interparticle chemical bonds to form [71].
The following workflow diagram summarizes the decision-making process for selecting an appropriate anti-agglomeration strategy.
The Scientist's Toolkit: Essential Reagents and Materials
Successful dispersion requires a carefully selected toolkit of reagents and materials. The following table catalogues key items and their functions.
Table 2: Essential Research Reagents for Nanofiller Dispersion
| Reagent/Material | Function/Principle | Example Applications |
|---|---|---|
| Polyethyleneimine (PEI) | A polymer wetting/binding agent; provides steric hindrance and can functionalize surfaces. | Dispersing nanodiamonds in Pebax matrix [73]. |
| Silane Coupling Agents | Forms chemical bridges between inorganic filler and organic polymer matrix. | Improving interfacial adhesion in epoxy/silica composites. |
| Citric Acid | Anionic surfactant; provides electrostatic stabilization to nanoparticles. | Stabilizing nano-gold and nano-palladium powders [70]. |
| Polyvinylpyrrolidone (PVP) | Non-ionic polymer dispersant; acts as a steric stabilizer. | Coating SiO₂ nanoparticles to improve compatibility [71]. |
| Sodium Polyphosphate | Inorganic electrolyte dispersant; increases surface potential for electrostatic repulsion. | Aqueous dispersion of various oxide nanoparticles [70]. |
| n-Butanol | Solvent for azeotropic distillation; reduces capillary force and replaces surface -OH groups. | Preventing hard agglomeration in gel-derived oxides [71]. |
| Graphene Oxide (GO) | 2D carbon nanofiller with functionalizable surface. | Major reinforcing phase in nacre-mimetic composites [72]. |
| Hexagonal Boron Nitride (hBN) | 2D nanosheet; used synergistically to prevent agglomeration of other fillers. | Hybrid filler with GO in PVA composites [72]. |
Quantitative Comparison of Dispersion Performance
Evaluating the effectiveness of a dispersion strategy often involves measuring the resultant composite's properties. The following table compares the mechanical outcomes of different strategies as reported in the literature.
Table 3: Mechanical Property Enhancement from Different Dispersion Strategies
| Dispersion Strategy | Filler/Matrix System | Filler Content | Key Improvement | Reference |
|---|---|---|---|---|
| PEI Surface Modification | Nanodiamond / Pebax | 0.1 - 1.5 wt% | Improved CO₂/N₂ selectivity and filler dispersion in MMMs. | [73] |
| Hybrid GO-hBN Filler | GO-hBN / PVA | 80 wt% | 787% increase in Young's Modulus vs. pure PVA. | [72] |
| Hybrid GO-hBN Filler | GO-hBN / PVA | 10 wt% | 106% increase in Tensile Strength vs. pure PVA. | [72] |
| Silica Nanoparticle Addition | Silica / Polyimide | 3 wt% | 39% improvement in transverse Young's modulus of piezoelectric nanocomposites. | [7] |
The "dispersion dilemma" in polymer nanocomposites is a complex but surmountable challenge. No single strategy is universally applicable; the choice depends on the specific nanofiller, polymer matrix, target loading, and desired properties. Chemical modification provides robust, thermodynamic solutions by altering surface chemistry. Physical processing offers direct, mechanical means to break apart agglomerates. For the demanding goal of high filler loading, innovative approaches like hybrid filler systems demonstrate remarkable efficacy by leveraging synergistic effects. The protocols and data summarized herein provide a foundational toolkit for researchers to design effective dispersion pathways, which is a prerequisite for unlocking the full potential of polymer nanocomposites in advanced applications.
The integration of polymer nanocomposites into biomedical devices, from bone scaffolds to drug delivery systems, hinges on a critical step: comprehensive biocompatibility and toxicity assessment. Within the broader context of polymer nanocomposites fabrication research, establishing a robust biological safety profile is not merely a regulatory hurdle but a fundamental component of the development process. These advanced materials, which combine polymers with nanoscale fillers like bioactive glass or metals, present unique interactions with biological systems that must be thoroughly evaluated [74] [4]. This document provides detailed application notes and experimental protocols, aligned with international standards such as ISO 10993, to guide researchers and scientists through the essential testing strategies for ensuring the safety of polymer nanocomposites in biomedical applications [75] [76].
Foundational Principles of Biocompatibility Assessment
Biocompatibility is not an intrinsic property of a material but is defined within the context of a specific medical device's application. The U.S. Food and Drug Administration (FDA) and other international regulatory bodies evaluate the biocompatibility of a device in its "final finished form," considering the interactions between all component materials, manufacturing processes (including sterilization), and the intended clinical use [75]. The assessment is guided by the nature, frequency, and duration of the device's contact with the body [75]. The core of this evaluation for nearly all medical devices rests on the "Big Three" tests: cytotoxicity, sensitization, and irritation [76]. A risk-based approach is mandated, where existing scientific data and in vitro studies should be leveraged to reduce the need for animal testing, in alignment with the 3Rs (Replacement, Reduction, and Refinement) principles [76].
Core Testing Protocols for Polymer Nanocomposites
The following protocols are essential for the initial biological safety evaluation of polymer nanocomposite-based devices. Testing is typically performed using extracts of the device material, prepared according to ISO 10993-12, using solvents like physiological saline or cell culture medium to simulate the release of leachable substances under clinical conditions [76] [77].
Cytotoxicity Testing (The Big Three)
Purpose: To determine if the nanocomposite releases substances that are toxic to living cells, providing an initial screening for acute toxicity [76] [77].
In Vitro Quantitative Assay (MTT Assay)
- Principle: This colorimetric method measures the reduction of yellow MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) to purple formazan by mitochondrial succinate dehydrogenase in living cells [77].
- Protocol:
- Cell Culture: Seed mammalian cells (e.g., L929 mouse fibroblasts) in a 96-well microplate and incubate until a monolayer forms.
- Sample Preparation: Prepare an extract of the polymer nanocomposite by immersing it in cell culture medium under standardized conditions (e.g., 24-72 hours at 37°C). A neat extract (undiluted) is typically tested first [76].
- Exposure: Replace the culture medium in the wells with the prepared extract. Include control wells with culture medium only and a known cytotoxic substance as a positive control.
- Incubation: Incubate the plates for 24 hours at 37°C with 5% CO₂.
- Viability Measurement: Add the MTT reagent to each well and incubate further. Living cells will convert MTT to formazan. Solubilize the formed crystals and measure the absorbance using a microplate reader (e.g., Bio-Tek ELx808) [77].
- Data Analysis: Calculate the percentage of cell viability relative to the negative control. A cell viability of ≥70% is generally considered a non-cytotoxic response, though acceptance criteria should be defined based on the device's intended use [76].
Other Methods: Qualitative assays like the Direct Contact or Agar Diffusion tests are used for screening, but quantitative evaluation is preferable per regulatory guidance [77].
Sensitization Testing (The Big Three)
Purpose: To evaluate the potential of the nanocomposite to cause an allergic or hypersensitivity reaction upon repeated or prolonged exposure [77].
- Murine Local Lymph Node Assay (LLNA)
- Principle: This test quantifies the proliferation of T-lymphocytes in the lymph nodes draining the application site in response to a sensitizer. It is preferred from an animal welfare perspective [77].
- Protocol:
- Test Material: Use an extract of the nanocomposite or specific chemicals leached from it.
- Animal Model: Mice (typically young adult females) are used.
- Dosing: Apply the test extract topically to the dorsum of both ears daily for three consecutive days.
- Lymphocyte Proliferation: Several days after the final application, inject the mice with radiolabeled thymidine. After a few hours, excise the draining lymph nodes and create a single-cell suspension.
- Measurement: The incorporation of radiolabeled thymidine into the DNA of proliferating lymphocytes is measured via beta-scintillation counting.
- Data Analysis: A test material is considered a sensitizer if it induces a three-fold or greater increase in lymphocyte proliferation compared to the vehicle control group [77].
Irritation Testing (The Big Three)
Purpose: To estimate the potential of the nanocomposite to cause localized irritation (redness, swelling) at the site of contact [77].
- Intracutaneous Test
- Principle: This test is recommended for devices with internal contact and reliably detects irritation due to extractable chemicals [77].
- Protocol:
- Test Material: Prepare extracts of the nanocomposite using both polar (e.g., saline) and non-polar (e.g., vegetable oil) solvents.
- Animal Model: Albino rabbits are typically used.
- Injection: Intradermally inject 0.2 mL of each extract, along with control blanks, at multiple sites on the rabbit's back.
- Observation: Observe the injection sites for evidence of erythema (redness), edema (swelling), and other toxic signs at 24, 48, and 72 hours after injection.
- Scoring: Score the reactions against a standardized scale. The mean scores for the test material should not significantly exceed those for the control blanks [77].
Quantitative Data and Material Characterization
For polymer nanocomposites, biological testing must be correlated with material properties to understand structure-function relationships. The table below summarizes key quantitative data from a study on Polylactic Acid/Nanobioglass (PLA/n-BG) composites, illustrating how nanofiller incorporation influences properties.
Table 1: Quantitative Properties of PLA/Nanobioglass (n-BG) Composites for Biomedical Applications [74]
| Material Property | Neat PLA | PLA with 5 wt.% n-BG | PLA with 10 wt.% n-BG | Testing Method / Notes |
|---|---|---|---|---|
| Elastic Modulus | 1.49 ± 0.44 MPa | 2.85 ± 0.99 MPa | Data not specified | Mechanical testing. A 91.3% increase with 5% n-BG. |
| Cell Viability (HeLa cells) | >80% (Baseline) | >80% | >80% | MTT or similar assay. Demonstrated non-cytotoxicity. |
| Crystallinity | 7.1% | 4.98% | Data not specified | Differential Scanning Calorimetry (DSC). |
| Glass Transition Temp. (Tg) | 53 °C | 63 °C | Data not specified | Differential Scanning Calorimetry (DSC). |
| Antimicrobial Efficacy | Not applicable | Effective | Effective | n-BGs alone inhibited growth of E. coli, S. aureus, etc., at 20 w/v%. |
Experimental Workflow for Biocompatibility Assessment
The following diagram outlines a logical, risk-managed testing workflow for a polymer nanocomposite medical device, from material understanding to final testing decisions.
Diagram 1: Biocompatibility testing workflow for medical devices.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful biocompatibility testing relies on a suite of well-defined reagents and materials. The following table details key items used in the protocols described herein.
Table 2: Essential Research Reagent Solutions for Biocompatibility Testing
| Reagent / Material | Function / Description | Application in Protocols |
|---|---|---|
| L929 Mouse Fibroblasts | A standard, well-characterized mammalian cell line used for cytotoxicity testing. | Cytotoxicity (MTT) Assay [76] |
| MTT Reagent | (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide); a yellow tetrazole reduced to purple formazan by living cells. | Quantitative measurement of cell viability in the MTT Assay [77] |
| Extraction Solvents | Polar and non-polar liquids used to simulate the leaching of substances from a material. Common solvents include Sodium Chloride (0.9%) and Vegetable Oil. | Preparation of device/material extracts for cytotoxicity, sensitization, and irritation tests [76] [77] |
| Complete Freund's Adjuvant (CFA) | An immunopotentiator used to enhance the skin sensitization response. | Guinea Pig Maximization Test (GPMT) for sensitization [77] |
| Radio-labeled Thymidine | A nucleotide incorporated into DNA during replication; used to measure cell proliferation. | Quantification of lymphocyte proliferation in the Murine Local Lymph Node Assay (LLNA) [77] |
| Polymer Nanocomposite Extract | The test article itself, prepared by exposing the final finished device to an extraction solvent under controlled conditions. | The central test sample for all in vitro and in vivo biocompatibility tests [75] [76] |
The translation of polymer nanocomposites (PNCs) from a laboratory discovery to a clinically viable product is predominantly hindered by the challenges of scalable manufacturing and batch-to-batch consistency. Achieving a uniform dispersion of nanofillers within a polymer matrix is a fundamental thermodynamic and kinetic challenge that, if not overcome, leads to highly variable material properties and performance [24] [78]. This application note details advanced fabrication paradigms and provides standardized protocols designed to bridge this critical lab-to-clinic gap, enabling the reproducible production of high-performance PNCs for biomedical applications such as tissue engineering and drug delivery [78] [66].
Performance Data of Advanced PNC Fabrication Strategies
The table below summarizes quantitative data and key characteristics of modern fabrication approaches that enhance scalability and consistency for biomedical PNCs.
Table 1: Quantitative Performance and Characteristics of Advanced PNC Fabrication Strategies
| Fabrication Strategy | Reported Mechanical Enhancement | Key Consistency/Scalability Advantage | Relevant Nanofiller Types | Primary Biomedical Application |
|---|---|---|---|---|
| "Create-instead-of-Add" (Fibrillation) | Tensile strength & modulus increase of 300-400% vs. conventional blends [24] | Bypasses thermodynamic dispersion hurdles; creates uniform nanofibrils in-situ [24] | Polymer-polymer blends | Polymer-polymer nanocomposites, scaffolds |
| Relaxation-Enhanced with Bound Loops | Maintains fluid-like dynamics even at high NP-loading; enhances glassy state toughness and strength [23] | Dynamic, loose particle network ensures consistent melt processability (e.g., for extrusion) [23] | Silica nanoparticles, metal nanoparticles | Processable high-loading composites for devices |
| Porous Nanoparticle Reinforcement | >20% enhancement in Young's modulus even with weak interfacial interactions [79] | Unique structural confinement effect reduces chain mobility, improving consistency [79] | Porous silica, porous metal oxides | High-stiffness and damping composites for implants |
| In Situ Polymerization | Strong interface between matrix and nanoparticles; enhanced electrical/thermal properties [66] [80] | Prevents nanoparticle agglomeration by building polymer around fillers [80] | Carbon nanotubes, graphene, metal nanoparticles | Conductive scaffolds, drug delivery systems |
| Twin-Screw Extrusion | Continuous, industrial-scale production; uniform dispersion of nanoparticles [80] | Industry-standard, continuous process; applicable to thermoplastics [80] | Nanoclays, carbon nanotubes, nanosilica | Biodegradable implants, packaging |
Detailed Experimental Protocols
Protocol: Fabrication of Relaxation-Enhanced PNCs via Bound Polymer Loops
This protocol details the molecular design of PNCs with enhanced processability and mechanical properties by introducing bound polymer loops on nanoparticle surfaces, mitigating the formation of a rigid particle network [23].
3.1.1. Workflow Visualization
3.1.2. Materials and Reagents
- Silica Nanoparticles (SiOx-NPs): 65 ± 10 nm diameter, as the core reinforcing filler [23].
- Poly(styrene-ran-4-hydroxystyrene) [P(S-ran-HS)]: Statistical copolymer; the 4-hydroxystyrene (HS) units provide strong H-bonding anchors to the silica surface [23].
- Polystyrene (PS) Matrix: ( M_w ) = 370 kg mol(^{-1}), PDI = 1.07, as the bulk composite material [23].
- Methyl Ethyl Ketone (MEK) & Toluene: Solvents for casting and mixing composite dispersions [23].
- Chloroform: Solvent for leaching non-attached polymer chains [23].
3.1.3. Step-by-Step Procedure
- Polymer Adsorption: Disperse silica NPs in a P(S-ran-HS) matrix by casting a composite dispersion in methyl ethyl ketone. Dry the cast dispersion to form a solid composite [23].
- Thermal Annealing for Binding: Anneal the composite at 150°C (Tg + 50°C) for 24 hours under vacuum. This step expedites the adsorption and pinning of the HS components in the copolymer chains onto the NP surface [23].
- Solvent Leaching to Create Bound Loops (BL): Wash the annealed composite with chloroform to remove all non-attached P(S-ran-HS) chains. This yields silica NPs covered with a layer of bound polymer loops (BL–SiOx NPs) [23].
- BL Layer Characterization: Confirm the formation and thickness of the bound loop layer using Transmission Electron Microscopy (TEM), Thermogravimetric Analysis (TGA), and Atomic Force Microscopy (AFM). The loop thickness can be controlled by the HS mole fraction ((f_{HS})) in the copolymer [23].
- Composite Fabrication: Mix the obtained BL–SiOx NPs with a toluene solution of PS. Allow the mixture to dry naturally at room temperature to form the final composite [23].
- Final Processing: Subject the composite to a final thermal annealing step at Tg + 30°C to remove any residual solvents [23].
Protocol: In Situ Fibrillation for "Genuine" Polymer-Polymer Nanocomposites
This protocol describes a "create-instead-of-add" method to generate genuine nanocomposites by in-situ formation of nanofibrils, completely bypassing the challenges of dispersing pre-made nanofillers [24].
3.2.1. Workflow Visualization
3.2.2. Materials and Reagents
- Immiscible Polymer Blends: A two-component system where the minor phase will form the fibrillar reinforcement [24].
- Selective Solvent: A chemical that can selectively dissolve and extract the matrix polymer without affecting the nanofibrils [24].
3.2.3. Step-by-Step Procedure
- Blend Preparation: Begin with an immiscible blend of two polymers [24].
- Cold Drawing: Subject the polymer blend to a cold drawing process. This mechanical deformation transforms the minor component of the blend into uniformly dispersed nanofibrils within the matrix, resulting in a nanofibrillar polymer-polymer composite [24].
- Optional: Nanofibril Isolation (for SPCs): To create neat nanofibrils, selectively extract the matrix component from the cold-drawn blend using an appropriate solvent [24].
- Optional: Compression Molding: Convert the isolated neat nanofibrils into a nanofibrillar single-polymer composite (SPC) via compression molding [24].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Fabricating Consistent Polymer Nanocomposites
| Reagent/Material | Function in Fabrication | Key Consideration for Scalability |
|---|---|---|
| Poly(styrene-ran-4-hydroxystyrene) | Creates bound polymer loops on NP surfaces to enhance interfacial relaxation and processability [23]. | The mole fraction of HS ((f_{HS})) precisely controls bound loop thickness, a critical consistency parameter [23]. |
| Porous Silica Nanoparticles | Zero-dimensional nanofiller with unique surface configuration for mechanical reinforcement [79]. | Porous surface provides structural confinement and enhanced interfacial bonding, overcoming stiffness-damping trade-off without requiring high loadings [79]. |
| Surface-Modified Nanoclays (e.g., Montmorillonite) | Plate-like nanofiller for improving mechanical strength and barrier properties [3]. | Organo-modification is essential for compatibilization with polymer matrices; dispersion quality is critical [80] [3]. |
| Multi-Walled Carbon Nanotubes (MWCNTs) | One-dimensional nanofiller for enhancing electrical conductivity and mechanical strength [3] [79]. | Prone to agglomeration; require advanced dispersion techniques (e.g., twin-screw extrusion, ultrasonication) [80]. |
| Twin-Screw Extruder | Continuous mechanical compounding equipment for dispersing nanofillers in molten polymers [80]. | Industry-standard for scalable production; screw design and shear profiles must be optimized for specific nanofiller types [80]. |
| Solvents for Leaching (e.g., Chloroform) | Selective removal of polymer components to isolate nanostructures (e.g., bound loops, nanofibrils) [24] [23]. | Solvent choice, volume, and contact time must be standardized to ensure reproducible extraction without damaging the desired nanostructure. |
The escalating global environmental crisis, driven significantly by the accumulation of non-biodegradable petroleum-based plastics, has intensified the search for sustainable material alternatives [81]. This push for sustainability has accelerated the shift towards green polymer nanocomposites (GPNCs)—hybrid materials that combine biodegradable or bio-based polymer matrices with eco-friendly nanoscale reinforcements [81]. These composites are engineered to match or exceed the performance of conventional plastics while drastically reducing environmental impact, supporting global initiatives like the European Green Deal and the United Nations Sustainable Development Goals [81]. GPNCs represent a convergence of material innovation and environmental stewardship, finding relevance across a diverse range of high-impact sectors including packaging, automotive, aerospace, water treatment, and biomedical devices [81] [62]. This document provides detailed application notes and experimental protocols for the fabrication of these advanced materials, framed within the broader context of research on polymer nanocomposites fabrication methods.
Core Material Components: A Research Toolkit
The performance of green polymer nanocomposites is dictated by the synergistic relationship between their two primary components: the biopolymer matrix and the nanofiller reinforcement.
Green Polymer Matrices
The matrix forms the continuous phase of the composite, largely determining its basic processing, biodegradability, and biocompatibility. The tables below catalog the primary natural and synthetic biopolymers used in GPNCs.
Table 1: Natural Biopolymer Matrices
| Biopolymer | Source | Key Properties | Inherent Limitations | Primary Research Applications |
|---|---|---|---|---|
| Starch [81] | Wheat, corn, potatoes | Thermoplastic, affordable, biodegradable | Brittle, high water sensitivity | Biocomposites, packaging films |
| Cellulose [81] [82] | Plant biomass (wood, husks) | Abundant, high stiffness, biodegradable | High crystallinity, difficult to process | Films, reinforcement via CNC/CNF |
| Chitosan [81] [82] | Crustacean shells (chitin) | Biocompatible, antimicrobial, film-forming | Poor mechanical strength, water sensitive | Wound dressings, drug delivery, packaging |
| Alginate [81] [82] | Brown seaweed | Ionic gelation, biocompatible, hydrophilic | Mechanically weak, variable structure | Tissue engineering, encapsulation, packaging |
Table 2: Bio-based Synthetic Polymer Matrices
| Biopolymer | Feedstock | Key Properties | Inherent Limitations | Primary Research Applications |
|---|---|---|---|---|
| PLA (Polylactic Acid) [81] [22] | Corn sugar, sugarcane | Transparent, compostable, high modulus | Brittle, low impact strength, poor barrier | 3D printing, packaging, textiles |
| PHA (Polyhydroxyalkanoates) [81] [22] | Microbial fermentation | Biodegradable, biocompatible, versatile | High cost, variable properties | Biomedical implants, packaging |
| PBS (Polybutylene Succinate) [81] | Bio-succinic acid | Ductile, good processability | Low melt strength, moderate barrier | Flexible packaging, mulch films |
| PCL (Polycaprolactone) [81] | Petrochemical (but biodegradable) | Highly flexible, low melting point | Low strength, slow degradation | Drug delivery, soft tissue scaffolds |
Eco-Friendly Nanofillers
Nanofillers are incorporated at low loadings (typically 1-5 wt%) to enhance the mechanical, thermal, and functional properties of the biopolymer matrix. Their high surface area-to-volume ratio is key to their effectiveness.
Table 3: Classification and Properties of Eco-Friendly Nanofillers
| Nanofiller Category | Specific Examples | Key Morphology | Primary Property Enhancements | Research Applications |
|---|---|---|---|---|
| Clay-Based [81] [83] [3] | Montmorillonite, Halloysite, Cloisite 30B | Layered silicate (nanoplatelets) | Mechanical strength, barrier properties, flame retardancy | Packaging, automotive parts |
| Carbon-Based [83] [3] | Cellulose Nanocrystals (CNC), Graphene Oxide, CNTs | Nanotubes, sheets, crystals | Electrical conductivity, tensile strength, thermal stability | Sensors, energy storage, composites |
| Metal/Metal Oxide [81] [62] [3] | Silver (Ag), Zinc Oxide (ZnO), Titanium Dioxide (TiO2) | Spherical/rod nanoparticles | Antimicrobial, photocatalytic, UV blocking | Active packaging, wound dressings, coatings |
| Bio-derived [81] [83] | Biochar, Lignin Nanoparticles | Particulates | Reinforcement, sustainability, reduced cost | Green composites, waste utilization |
Diagram 1: GPNC Component Classification
Quantitative Performance Enhancements
The incorporation of nanofillers leads to measurable improvements in the properties of biopolymers. The following table summarizes key quantitative findings from recent research, providing benchmarks for material design.
Table 4: Documented Enhancement of Biopolymer Properties via Nanofillers
| Biopolymer Matrix | Nanofiller | Filler Loading | Key Property Enhancement | Source/Reference |
|---|---|---|---|---|
| Cellulose-Vinyl Ester [83] | Nanoclay (Cloisite 30B) | Not Specified | Significant reduction in water absorption | [83] |
| PLA (3D Printed) [7] | Silica Nanoparticles | 3 wt% | 39% improvement in transverse Young's modulus | [7] |
| General Biopolymers [81] | Functionalized Nanofillers | Not Specified | ~60-70% increase in modulus; 200% increase in elongation at break | [81] |
| General Biopolymers [81] | Metal Nanoparticles (e.g., Ag, ZnO) | Not Specified | Reduction in cell viability to <10%; 100% photocatalytic efficiency | [81] |
| General GPCs [22] | Natural Fibers & Nanomaterials | Not Specified | Up to 30% increase in tensile strength; 40% reduction in carbon footprint vs. conventional composites | [22] |
Detailed Experimental Protocols
This section outlines standardized protocols for the fabrication of green polymer nanocomposites, suitable for replication in a research setting.
Protocol 4.1: Solution Casting for Film Fabrication
Application Note: This method is ideal for laboratory-scale production of thin nanocomposite films, especially for packaging, biomedical membrane, and sensor applications [81] [22]. It is particularly suitable for heat-sensitive biopolymers like chitosan and starch.
Materials & Equipment:
- Biopolymer: e.g., Chitosan (medium molecular weight).
- Nanofiller: e.g., Cellulose Nanocrystals (CNC) suspension.
- Solvent: Aqueous acetic acid solution (1% v/v).
- Equipment: Magnetic stirrer/hotplate, ultrasonication bath, vacuum desiccator, glass petri dishes, precision balance.
Step-by-Step Procedure:
- Solution Preparation:
- Dissolve 1.0 g of chitosan in 100 mL of aqueous acetic acid solution (1% v/v) under constant magnetic stirring at 400 rpm for 6 hours at room temperature until fully dissolved to form a 1% w/v solution.
- Nanofiller Dispersion:
- Take 50 mL of the prepared chitosan solution. Add the desired mass of CNC (e.g., 3 wt% relative to chitosan) dropwise under vigorous stirring.
- Transfer the mixture to an ultrasonication bath and sonicate for 30 minutes at 40% amplitude with pulse cycles (10 s on, 5 s off) to ensure homogeneous dispersion and prevent agglomeration.
- Casting and Drying:
- Pour the final mixture into a clean, level glass petri dish.
- Allow the solvent to evaporate at ambient conditions for 24-48 hours, covered with a breathable cloth to prevent dust contamination.
- Alternatively, for faster drying, place the cast film in a vacuum oven at 40°C for 12 hours.
- Post-Processing:
- Carefully peel the resulting film from the petri dish.
- Condition the film in a controlled environment (e.g., 50% relative humidity, 25°C) for at least 24 hours before further testing and characterization.
Protocol 4.2: Melt Blending and Compression Molding
Application Note: This solvent-free, scalable method is compatible with thermoplastic biopolymers like PLA and PHA and is common in industrial processes for creating structural components [22] [7].
Materials & Equipment:
- Biopolymer: e.g., PLA pellets.
- Nanofiller: e.g., surface-modified Montmorillonite nanoclay.
- Equipment: Twin-screw micro-compounder or internal mixer, compression molding press, Teflon sheets or release agent.
Step-by-Step Procedure:
- Pre-drying:
- Dry PLA pellets and the nanofiller in a vacuum oven at 60°C for at least 12 hours to remove moisture.
- Melt Blending:
- Manually pre-mix the dried PLA pellets with the nanofiller (e.g., 5 wt%) to form a coarse blend.
- Feed the mixture into a pre-heated twin-screw micro-compounder. Set the temperature profile along the barrels to 175-185-190-180°C. Maintain a screw speed of 100 rpm for 5 minutes to ensure sufficient shear for dispersion and distributive mixing.
- Compression Molding:
- Transfer the extruded melt immediately into a pre-heated mold in a compression press.
- Compress at 180°C under 5 MPa pressure for 3 minutes, followed by 10 MPa for 2 minutes to ensure complete flow and consolidation.
- Cool the mold under pressure using the press's cooling system to room temperature to form a plaque of defined thickness (e.g., 1 mm).
Protocol 4.3: In Situ Polymerization
Application Note: This technique offers the highest level of nanofiller dispersion by performing the polymerization reaction in the presence of the pre-dispersed nanofiller, leading to superior mechanical and thermal properties [81] [7]. It is used for creating high-performance nanocomposites.
Materials & Equipment:
- Monomer: e.g., Lactide.
- Nanofiller: e.g., Graphene Oxide (GO) sheets.
- Catalyst: e.g., Tin(II) octoate.
- Solvent: Toluene.
- Equipment: Schlenk flask, magnetic stirrer, oil bath, inert gas (N₂ or Ar) supply.
Step-by-Step Procedure:
- Nanofiller Pre-dispersion:
- Disperse a precise amount of GO (e.g., 0.5 wt%) in 50 mL of anhydrous toluene using probe ultrasonication for 1 hour under an inert atmosphere.
- Reaction Setup:
- Transfer the GO dispersion to a clean, dry Schlenk flask equipped with a magnetic stir bar.
- Add purified lactide monomer to the flask at a monomer-to-nanofiller ratio designed to yield the target final composite composition.
- Polymerization:
- Flush the flask with inert gas and place it in an oil bath pre-heated to 130°C.
- Using a gas-tight syringe, inject the catalyst (Tin(II) octoate, 0.025 wt% relative to monomer) into the reaction mixture.
- Allow the reaction to proceed under constant stirring for 6-12 hours.
- Purification:
- Terminate the reaction by cooling the flask to room temperature.
- Precipitate the resulting PLA/GO nanocomposite into cold methanol, then filter and wash several times to remove any unreacted monomer and catalyst.
- Dry the final product under vacuum at 50°C to constant weight.
Diagram 2: GPNC Fabrication Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Reagent Solutions for GPNC Formulation
| Reagent / Material | Function / Role | Example & Notes |
|---|---|---|
| Silane Coupling Agents [22] [83] | Improve interfacial adhesion between hydrophilic nanofillers and hydrophobic polymers. | (3-Aminopropyl)triethoxysilane (APTES) is widely used to functionalize nanoclay and silica nanoparticles. |
| Citric Acid / Acetic Acid [82] | Solvent and catalyst for biopolymer processing. | Aqueous Acetic Acid (1-2% v/v) is the standard solvent for dissolving Chitosan. |
| Tin(II) Octoate [81] | Catalyst for ring-opening polymerization of esters. | Standard catalyst for synthesizing PLA via lactide polymerization; requires strict anhydrous conditions. |
| Glutaraldehyde | Crosslinking agent to enhance mechanical strength and water resistance. | Can crosslink chitosan films; concentration must be optimized to maintain biocompatibility. |
| Surfactants (e.g., CTAB) [83] | Aid in the dispersion and exfoliation of nanofillers within the matrix. | Cetyltrimethylammonium bromide (CTAB) can be used to modify the surface of layered silicates. |
In the rapidly advancing field of polymer nanocomposites (PNCs), the bridge between laboratory-scale innovation and industrial-scale application is built upon rigorous process parameter optimization. The enhanced electrical, mechanical, and thermal properties of PNCs—attributes that make them indispensable in applications from biomedical devices to flexible electronics—are not merely a function of their constituent materials but are profoundly dictated by the fabrication process itself [4] [84]. Achieving reproducible, high-quality output requires a systematic approach to fine-tuning a multitude of interdependent parameters, a challenge that grows in complexity with the adoption of advanced manufacturing techniques like additive manufacturing [35] [85]. This application note provides a structured framework and detailed protocols for optimizing key fabrication processes to ensure consistent performance and accelerate the development of next-generation PNCs.
Critical Process Parameters by Fabrication Technique
The optimal properties of a polymer nanocomposite are locked in during fabrication. Unlocking them requires precise control over technique-specific parameters, which directly influence filler dispersion, interfacial adhesion, and ultimate structural integrity.
Electrospinning of Piezoelectric Fibers
Electrospinning is a versatile technique for preparing polymer nanocomposite fibres, notably from polyvinylidene fluoride (PVDF) and its copolymers, for piezoelectric applications. It offers advantages such as a large specific surface area, controllable structure, and ease of fabrication [86]. The critical parameters governing fibre morphology and piezoelectric performance are summarized in Table 1.
Table 1: Key Parameter Optimization for Electrospinning PNCs
| Parameter Category | Specific Parameter | Optimized Value/Range | Impact on Output Quality |
|---|---|---|---|
| Solution Properties | Polymer Concentration | 10-20 wt% (system-dependent) | Determines fiber continuity; low concentration causes bead formation, high concentration increases fiber diameter. |
| Solvent Volatility | High (e.g., DMF/Acetone mixes) | Facilitates rapid solvent evaporation and solid fiber formation. | |
| Filler Dispersion | Uniform via sonication/shear mixing [85] | Prevents nanoparticle agglomeration, ensures homogeneous properties. | |
| Process Parameters | Applied Voltage | 10-25 kV | Governs jet initiation and stretching; too low fails to form a Taylor cone, too high causes instability. |
| Flow Rate | 0.5-3 mL/h | Controls fiber diameter; high rate leads to thick, wet fibers. | |
| Collector Distance | 10-20 cm | Allows sufficient time for solvent evaporation; short distance results in wet, fused fibers. | |
| Environmental | Ambient Humidity | Controlled (20-50%) | High humidity can cause pore formation or fiber collection issues. |
Material Extrusion Additive Manufacturing
Material extrusion (MEX), including Fused Filament Fabrication (FFF) and Direct Ink Writing (DIW), is a transformative approach for fabricating complex PNC geometries. Parameter optimization is critical to minimize defects like voids and weak interfacial bonding [85].
Table 2: Key Parameter Optimization for Material Extrusion of PNCs
| Parameter Category | Specific Parameter | Optimized Value/Range | Impact on Output Quality |
|---|---|---|---|
| Nozzle & Temperature | Nozzle Diameter | 0.2-0.8 mm (adapt to filler size) | Prevents clogging; smaller diameters improve resolution but risk clogging. |
| Nozzle Temperature | Material-dependent (e.g., ~200°C for PLA) | Ensures proper melt viscosity; too low increases pressure, too low causes poor layer adhesion. | |
| Bed Temperature | Material-dependent (e.g., 60°C for PLA) | Improves first-layer adhesion and reduces warping. | |
| Print Motion | Print Speed | 20-80 mm/s | Balances print time and quality; high speed can cause under-extrusion or layer shifting. |
| Layer Height | 0.1-0.3 mm | Affects resolution and strength; smaller height improves resolution but increases print time. | |
| Post-Processing | Thermal Annealing | Application-specific (e.g., 100°C for 1h) | Enhances crystallinity and inter-layer diffusion, improving mechanical properties [85]. |
Vat Photopolymerization
Vat Photopolymerization (VPP) methods like Stereolithography (SLA) and Digital Light Processing (DLP) are valued for their high resolution in fabricating PNCs. Key challenges include managing light scattering from fillers and maintaining resin homogeneity [85].
Table 3: Key Parameter Optimization for Vat Photopolymerization of PNCs
| Parameter Category | Specific Parameter | Optimized Value/Range | Impact on Output Quality |
|---|---|---|---|
| Resin Formulation | Filler Loading | <5 wt% for nanoparticles [85] | High loadings scatter light, leading to incomplete curing and poor mechanical properties. |
| Photoabsorber Concentration | Systematically optimized | Controls cure depth and prevents light penetration into subsequent layers. | |
| Curing Parameters | Light Wavelength | 365-405 nm (matches photoinitiator) | Must match the absorption peak of the photoinitiator for efficient curing. |
| Exposure Time/Light Intensity | Determined via working curve analysis | Insufficient exposure causes weak, tacky parts; excessive exposure causes over-curing and delamination. | |
| Post-Processing | Washing | In suitable solvent (e.g., isopropanol) | Removes uncured resin from the part surface. |
| Post-Curing | UV light & sometimes heat | Ensures complete conversion of the polymer matrix, enhancing cross-linking density and final properties [87]. |
Experimental Protocols for Process Optimization
Protocol: Systematic Optimization of Electrospinning Parameters for PVDF Nanofibers
Objective: To reproducibly fabricate bead-free, uniform PVDF nanofibers with high piezoelectric phase (β-phase) content.
Materials:
- Polymer: Polyvinylidene fluoride (PVDF) pellets.
- Solvent: A binary solvent system of N,N-Dimethylformamide (DMF) and Acetone.
- Equipment: Standard electrospinning apparatus with a high-voltage power supply, syringe pump, and grounded collector.
Procedure:
- Solution Preparation: Prepare a 15 wt% PVDF solution in a 7:3 (v/v) DMF/Acetone solvent mixture. Stir at 60°C for 6 hours until a clear, homogeneous solution is obtained.
- Parameter Screening: Load the solution into a syringe. Systemically vary one parameter at a time while holding others constant:
- Flow Rate: Test 0.5, 1.0, 1.5, and 2.0 mL/h.
- Applied Voltage: Test 15, 20, and 25 kV.
- Tip-to-Collector Distance (TCD): Test 12, 15, and 18 cm.
- Sample Collection: Collect fibers on aluminum foil for each parameter set. Label each sample clearly.
- Characterization & Analysis:
- Morphology: Analyze fiber diameter and bead density using Scanning Electron Microscopy (SEM).
- Crystallinity: Determine the β-phase content using Fourier-Transform Infrared Spectroscopy (FTIR) by calculating the ratio of the absorption peaks at 840 cm⁻¹ (β-phase) and 766 cm⁻¹ (α-phase).
- Validation: Once optimal parameters are identified (e.g., 1.0 mL/h, 20 kV, 15 cm TCD), run three independent replicates to confirm the reproducibility of fiber morphology and β-phase content.
Protocol: Optimizing FFF Printing for Short Carbon Fiber-Reinforced Composites
Objective: To determine the optimal printing parameters that maximize tensile strength and minimize void content in a short carbon fiber-reinforced thermoplastic (e.g., Nylon).
Materials:
- Filament: Short carbon fiber-reinforced thermoplastic filament (e.g., Nylon-CF).
- Equipment: FFF 3D printer with a hardened steel nozzle.
Procedure:
- Nozzle and Bed Setup: Install a 0.6 mm diameter hardened steel nozzle to mitigate abrasive wear from fibers. Apply a suitable adhesive (e.g., PVA glue) to the build plate to ensure adhesion.
- Printing Parameter Design: Print standardized tensile bars (per ASTM D638) while varying key parameters:
- Nozzle Temperature: Test 240°C, 250°C, and 260°C.
- Print Speed: Test 30 mm/s and 50 mm/s.
- Raster Angle: Test [0°/90°] and [+45°/-45°] patterns.
- Post-Processing: Anneal a subset of the printed tensile bars in an oven at 80°C for 4 hours to study the effect on inter-layer strength and crystallinity.
- Characterization & Analysis:
- Mechanical Testing: Perform tensile tests to determine ultimate tensile strength and Young's modulus.
- Structural Analysis: Examine the fracture surface and internal structure using SEM to assess fiber-matrix adhesion, void content, and failure mechanisms.
- Optimization: Use the data to identify the parameter set that yields the highest mechanical performance and lowest defect density.
FFF Process Optimization Workflow
The Scientist's Toolkit: Research Reagent Solutions
Successful fabrication of polymer nanocomposites relies on a suite of essential materials and reagents, each serving a critical function.
Table 4: Essential Materials for Polymer Nanocomposite Fabrication
| Material/Reagent | Function/Application | Key Considerations |
|---|---|---|
| PVDF (Polyvinylidene fluoride) | Piezoelectric polymer matrix for sensors and energy harvesting [86]. | High molecular weight grades promote better fiber formation during electrospinning. |
| Silver Nanoparticles (AgNPs) | Metallic nanofiller for antimicrobial activity and enhanced electrical conductivity [4] [84]. | Require surface functionalization for compatibility and to prevent agglomeration within the polymer matrix. |
| Carbon Nanofibers (CNFs) | Reinforcement filler to improve mechanical strength (toughness, modulus) and electrical conductivity [4] [87]. | Dispersion is critical; often require compatibilizers or surface oxidation for good matrix adhesion. |
| Sol-Gel Precursors (e.g., TEOS) | Used in in-situ synthesis of inorganic nanoparticles (e.g., silica) within a polymer matrix [4] [35]. | Offers precise control and high purity but the final product can be brittle, requiring careful aging and drying. |
| Photoinitiator (e.g., TPO-LAP) | Absorbs light to initiate polymerization in Vat Photopolymerization (SLA/DLP) resins [85]. | Concentration and compatibility with wavelength of light source are critical for achieving adequate cure depth and resolution. |
| Surface Modifying Agents (e.g., Silanes) | Coupling agents that improve the interfacial adhesion between inorganic nanofillers and the organic polymer matrix [4]. | Choice of agent depends on the chemistry of both the filler and the polymer. |
Advanced Workflow for Integrated Optimization
For complex systems, a holistic approach that integrates material selection with process parameters is necessary. This is particularly true for additive manufacturing, where design and fabrication are intrinsically linked.
Integrated PNC Development Workflow
Performance Validation and Technique Comparison: Selecting the Right Fabrication Route
Within the broader context of research on polymer nanocomposite fabrication, rigorous quality assurance is paramount. The properties of these advanced materials are profoundly influenced by their nanostructure, including the dispersion of nanofillers, interfacial interactions, and resulting morphological changes. This document provides detailed application notes and protocols for four cornerstone characterization techniques—X-ray Diffraction (XRD), Fourier-Transform Infrared Spectroscopy (FTIR), Scanning Electron Microscopy (SEM), and Thermal Analysis (TA)—framed specifically for quality assurance in polymer nanocomposites research. These techniques serve as critical tools for verifying successful fabrication, understanding structure-property relationships, and ensuring batch-to-batch consistency, which is especially relevant for high-value applications such as biomedical devices and drug delivery systems [4].
The following table summarizes the core quality assurance parameters measurable by each technique and their significance in evaluating polymer nanocomposites.
Table 1: Quality Assurance Characterization Techniques for Polymer Nanocomposites
| Technique | Core QA Parameters Measured | Significance in Polymer Nanocomposite Fabrication |
|---|---|---|
| XRD (X-Ray Diffraction) | Crystal structure, phase identification, crystallite size, interlayer spacing (d-spacing) in layered fillers [88]. | Confirms nanofiller exfoliation/intercalation in polymer matrix; monitors changes in polymer crystallinity due to nanofiller presence [89]. |
| FTIR (Fourier-Transform Infrared Spectroscopy) | Chemical functional groups, molecular structure, chemical bonding, interfacial interactions between polymer and nanofiller [88]. | Verifies successful surface modification of nanofillers; identifies covalent or hydrogen bonding at the polymer-filler interface [90]. |
| SEM (Scanning Electron Microscopy) | Surface morphology, nanofiller dispersion, agglomeration, fracture surface analysis, void content [88]. | Directly visualizes homogeneity of nanocomposite; identifies defects and agglomerates that compromise mechanical properties [88]. |
| Thermal Analysis (TGA/DSC) | Thermal stability (decomposition temperature), glass transition temperature (Tg), melting/crystallization behavior, composition percentage [89] [88]. | Assesses enhancement in thermal stability; reveals polymer chain mobility restrictions due to nanofiller interactions [89]. |
Experimental Protocols
Protocol: X-ray Diffraction (XRD) for Nanocomposite Structure
Principle: XRD determines the atomic and molecular structure of a crystal by measuring the angles and intensities of diffracted X-ray beams [88].
Materials:
- XRD Instrument: Powder X-ray diffractometer (e.g., Shimadzu 7000) [88].
- Sample Holder: Flat plate sample holder.
- Sample: Powdered nanocomposite or a flat, smooth film piece.
Procedure:
- Sample Preparation: For solid films, cut a piece to fit flush within the sample holder. For powders, pack the powder evenly into the holder to ensure a flat surface.
- Instrument Setup: Use Cu-Kα radiation (λ = 1.54056 Å). Set the scanning range (2θ) from 5° to 80° with a continuous scanning rate of 2° per minute [88].
- Data Collection: Load the sample into the diffractometer and initiate the scan.
- Data Analysis:
- Identify the characteristic diffraction peaks of the polymer matrix and the nanofiller.
- For layered nanofillers (e.g., clays), calculate the interlayer spacing (d-spacing) using Bragg's law:
nλ = 2d sinθ. An increase in d-spacing or disappearance of the filler's peak indicates intercalation or exfoliation, respectively [89]. - Estimate the average crystallite size of nanofillers using the Debye-Scherrer equation:
D = (0.9 × λ) / (β × cosθ), whereβis the full width at half maximum (FWHM) of the diffraction peak [88].
Protocol: Fourier-Transform Infrared (FTIR) Spectroscopy for Chemical Analysis
Principle: FTIR identifies chemical functional groups by measuring the absorption of infrared light at specific wavelengths, which correspond to molecular vibrations [91].
Materials:
- FTIR Spectrometer: (e.g., Shimadzu FTIR–Tracer 100) [88].
- Accessory: Attenuated Total Reflectance (ATR) crystal (e.g., diamond).
- Sample: Thin film or a small, solid piece of the nanocomposite.
Procedure:
- Background Collection: Place the ATR crystal in the spectrometer and collect a background spectrum with no sample.
- Sample Measurement: Place the nanocomposite film in direct contact with the ATR crystal. Apply consistent pressure to ensure good contact.
- Spectral Acquisition: Scan over a wavenumber range of 4000 to 400 cm⁻¹ [88].
- Data Analysis:
- Compare the spectrum of the nanocomposite with that of the pure polymer and the neat nanofiller.
- Identify shifts in the position of characteristic absorption bands (e.g., C=O, O-H, N-H), which indicate interfacial interactions like hydrogen bonding.
- Look for the appearance or disappearance of peaks to confirm successful chemical modification or covalent bonding.
Protocol: Scanning Electron Microscopy (SEM) for Morphology
Principle: SEM produces high-resolution images of a sample surface by scanning it with a focused beam of electrons and detecting secondary or backscattered electrons [88].
Materials:
- SEM Instrument: (e.g., Thermo Fisher Quattro ESEM) [88].
- Sample Stubs: Aluminum stubs with conductive adhesive tape.
- Sputter Coater: For applying a thin conductive layer (e.g., gold or carbon).
Procedure:
- Sample Preparation: Mount a small, representative piece of the nanocomposite on the stub using conductive tape. For brittle materials, analyze the fracture surface.
- Conductive Coating: Sputter-coat the sample with a 10-20 nm layer of gold to prevent charging under the electron beam.
- Imaging: Insert the sample into the SEM chamber. Select an accelerating voltage (e.g., 5-15 kV). Start at low magnification to locate the area of interest, then increase magnification to examine nanofiller dispersion, agglomeration, and polymer-filler interface.
- Data Analysis: Qualitatively assess the uniformity of nanofiller distribution. The presence of large, micron-sized agglomerates indicates poor dispersion and a failed fabrication process [88].
Protocol: Thermal Analysis (TGA & DSC)
Principle:
- TGA (Thermogravimetric Analysis): Measures mass change as a function of temperature, indicating thermal stability and composition [89] [92].
- DSC (Differential Scanning Calorimetry): Measures heat flow differences between a sample and reference, revealing thermal transitions like glass transition (Tg), melting, and crystallization [89].
Materials:
- TGA Instrument: (e.g., Shimadzu TGA-51) [88].
- DSC Instrument: (e.g., various commercial models) [89].
- Sample Crucibles: Standard alumina crucibles for TGA; sealed aluminum pans for DSC.
- Sample: 5-10 mg of finely cut or powdered nanocomposite.
TGA Procedure:
- Calibration: Calibrate the balance and temperature of the TGA.
- Loading: Place an empty reference crucible and the sample-containing crucible in the instrument.
- Experiment: Heat the sample from room temperature to 600-800 °C at a constant rate (e.g., 10 °C/min) under an inert nitrogen atmosphere [88].
- Data Analysis:
DSC Procedure:
- Calibration: Calibrate the DSC using indium or other standards for temperature and enthalpy.
- Loading: Hermetically seal the sample in an aluminum pan. Use an empty pan as a reference.
- Experiment: Run a heat-cool-heat cycle. Typically, heat from -50°C to 250°C (1st heat), cool back to -50°C, and heat again to 250°C (2nd heat), all at a rate of 10 °C/min [89].
- Data Analysis:
- From the second heating cycle, determine the glass transition temperature (Tg) as a midpoint of the heat capacity shift. An increase in Tg suggests restricted polymer chain mobility due to strong polymer-filler interactions [89].
- Analyze the melting temperature (Tm) and melting enthalpy (ΔHf) from the endothermic peak, and crystallization behavior from the cooling cycle.
Workflow and Relationships
The following diagram illustrates the integrated workflow of these techniques for comprehensive quality assurance in a single study, as demonstrated in recent research on PVA/SrTiO3/CNT nanocomposites [88].
Research Reagent Solutions
The following table lists essential materials and their functions for the fabrication and characterization of polymer nanocomposites, as referenced in the protocols.
Table 2: Essential Research Reagents and Materials for Nanocomposite Fabrication and Characterization
| Material/Reagent | Function/Application | Example Use Case |
|---|---|---|
| Polyvinyl Alcohol (PVA) | Polymer matrix; hydrophilic, non-toxic, high dielectric strength [88]. | Base material for synthesizing nanocomposite films via solution casting [88]. |
| Carbon Nanotubes (CNTs) | Nanofiller; enhances mechanical strength, electrical/thermal conductivity [89] [88]. | Used in mixtures with SrTiO₃ to improve thermal stability and optical properties of PVA films [88]. |
| Strontium Titanate (SrTiO₃) | Ceramic nanofiller; high dielectric constant, improves thermal and optical performance [88]. | Combined with CNTs as a hybrid dopant for PVA polymer films [88]. |
| Organically Modified Montmorillonite (OMMT) | Layered silicate nanofiller; improves barrier properties, thermal stability, and mechanical strength [89]. | Reinforcing phase in polyurethane and other polymer matrices to create intercalated/exfoliated structures [89]. |
| Conductive Coating (Gold) | Creates a conductive surface layer for non-conductive samples. | Essential for preventing charging during SEM imaging of polymer nanocomposites [88]. |
Polymer nanocomposites represent a revolutionary class of materials formed by dispersing nanoscale fillers into polymer matrices, yielding superior properties unattainable by their individual components. The fabrication methods employed critically determine the dispersion of nanofillers, the interfacial adhesion, and consequently, the final performance characteristics of the nanocomposite. This document provides a detailed comparative analysis of prevalent fabrication techniques, structured for researchers and scientists engaged in the development of advanced materials for pharmaceutical and biomedical applications. Within the broader context of thesis research on polymer nanocomposites, this analysis aims to bridge foundational knowledge with state-of-the-art protocols, empowering informed methodological selection.
Comparative Analysis of Fabrication Methods
The selection of an appropriate fabrication method is paramount, as it directly influences the nanofiller dispersion, the preservation of nanomaterial properties, and the ultimate functionality of the composite. The following sections and tables provide a quantitative and qualitative comparison of the most prominent techniques.
Table 1: Quantitative Comparison of Key Fabrication Methods
| Fabrication Method | Typical Nanofiller Loading (wt.%) | Processing Temperature | Scalability | Key Limitations |
|---|---|---|---|---|
| Melt Blending [93] [35] | 0.1 - 5 | High (Above polymer Tm) | Excellent (Industrial) | Potential nanofiller aggregation/breakage |
| Solution Blending [93] [35] | 0.5 - 5 | Low (Room Temp common) | Good | Solvent toxicity and removal issues |
| In-Situ Polymerization [93] [35] | 0.5 - 10 | Variable | Moderate | Complex synthesis, potential side reactions |
| Vapor-Phase Deposition [35] | N/A (Surface coating) | High | Moderate (Specialized) | Requires vacuum systems, high cost |
| Additive Manufacturing (3D Printing) [94] [35] | 0.5 - 5 | Variable (Depends on method) | Good (Rapidly improving) | Limited resolution, post-processing may be needed |
Table 2: Qualitative Comparison of Resultant Nanocomposite Properties
| Fabrication Method | Dispersion Quality | Interfacial Adhesion | Property Enhancement | Suitability for Biomedical Devices |
|---|---|---|---|---|
| Melt Blending | Moderate | Moderate | Good Mechanical, Thermal | Good (with biocompatible polymers) |
| Solution Blending | Good to Excellent | Good | Excellent Electrical, Optical | Moderate (Residual solvent concern) |
| In-Situ Polymerization | Excellent | Excellent | Superior Mechanical, Functional | Excellent for tailored drug delivery systems |
| Vapor-Phase Deposition | Excellent (as coating) | Excellent | Barrier, Surface Conductivity | Excellent for implantable sensor coatings |
| Additive Manufacturing | Moderate (Process-dependent) | Moderate (Layer adhesion) | High Design Freedom, Customization | Excellent for patient-specific scaffolds & devices |
Detailed Method Analysis
- Melt Blending: This method involves mixing nanofillers with a polymer melt, typically using a twin-screw extruder [95]. It is highly favored for its solvent-free nature and direct compatibility with large-scale industrial processes like extrusion and injection molding [93]. A key challenge is overcoming the strong van der Waals forces between nanofillers (e.g., ~300 nN/mm² for graphite exfoliation) within a highly viscous polymer melt, which can lead to agglomeration [93]. Twin-screw extruders are particularly effective due to their superior mixing capability, modular screw design, and precise control over parameters like shear force and temperature profile [95].
- Solution Blending: This technique relies on dispersing nanofillers and dissolving the polymer in a common solvent, followed by mixing and solvent removal [35]. It often achieves superior dispersion, especially for hydrophilic nanofillers like graphene oxide (GO) [93]. However, the process faces significant drawbacks, including the use of large quantities of toxic solvents, which pose health and environmental risks, and the difficulty of complete solvent removal, which can compromise material performance and biocompatibility [93].
- In-Situ Polymerization: In this approach, nanofillers are dispersed in a monomeric precursor, followed by polymerization. This allows for covalent bonding between the polymer matrix and the nanofiller surface, leading to excellent interfacial adhesion and potentially very high grafting densities [35]. While it can produce nanocomposites with outstanding properties, the process is often complex, can involve side reactions, and is less straightforward to scale than melt blending [93] [35].
- Additive Manufacturing (3D Printing): This represents a frontier in nanocomposite fabrication, enabling the creation of complex, custom geometries. Techniques like Fused Filament Fabrication (FFF)—which uses a pre-compounded nanocomposite filament—Vat Photopolymerization, and Powder Bed Fusion are being actively explored [94]. The primary challenges include ensuring uniform nanofiller dispersion in the feedstock and managing the influence of printing parameters on the final material's anisotropy and properties [94] [35].
Experimental Protocols
This section provides detailed, actionable protocols for two key fabrication methods relevant to drug development research: twin-screw extrusion and solution blending for film casting.
Protocol 1: Melt Compounding via Twin-Screw Extrusion
Application Note: This protocol is ideal for the high-throughput, solvent-free production of thermoplastic polymer nanocomposites (e.g., PLA, PP, PA12) with carbon-based nanofillers (GNPs, rGO) for applications requiring enhanced mechanical strength or electrical conductivity [93].
Materials:
- Polymer matrix (e.g., PLA pellets, dried at 80°C for 4 hours).
- Nanofiller (e.g., Graphene Nanoplatelets - GNP).
- Compatibilizer (if required; e.g., maleic anhydride grafted polymer).
Equipment:
- Co-rotating twin-screw extruder (L/D ratio ≥ 40).
- Gravimetric or volumetric feeder for polymer.
- Side-feeder or pre-mixing station for nanofiller.
- Water bath and pelletizer.
- Injection molding machine (for standard test specimen fabrication).
Procedure:
- Pre-Mixing: Pre-mix the dried polymer pellets with the nanofiller at the desired weight percentage (e.g., 0.5-3 wt.%) using a high-speed mixer to ensure an initial homogeneous dry blend.
- Extruder Setup: Configure the twin-screw extruder with a screw profile designed for high distributive and dispersive mixing. This typically incorporates kneading blocks and reverse elements. Set the temperature profile along the barrel according to the polymer's melting point and thermal stability. For PLA, a profile from 180°C (feed zone) to 200°C (die) is typical [93].
- Feeding & Compounding: Feed the pre-mixed material into the main hopper. If using a high nanofiller concentration, a side-feeder is recommended to introduce the filler downstream in the melting zone to reduce shear exposure. Set the screw speed between 200-500 rpm to achieve adequate shear for dispersion without excessive degradation.
- Strand Pelletizing: As the homogenized melt exits the die, guide the extruded strand through a water bath for cooling, and subsequently into a pelletizer to create uniform nanocomposite pellets.
- Specimen Preparation: Dry the pellets thoroughly (e.g., 80°C under vacuum) to remove absorbed moisture. Fabricate standard test specimens (e.g., ASTM tensile bars) using an injection molding machine with parameters optimized for the polymer nanocomposite.
Troubleshooting:
- Agglomeration: Increase the intensity of mixing elements (kneading blocks) in the screw profile or optimize the feed rate to screw speed ratio.
- Degradation: Lower the processing temperature profile or reduce the screw speed to minimize thermal and shear history.
- Poor Dispersion: Ensure the screw design has sufficient mixing sections and confirm the nanofiller is not being introduced too early in the melting process.
Protocol 2: Solution Blending and Film Casting
Application Note: This protocol is suited for producing thin, uniform nanocomposite films with excellent nanofiller dispersion, ideal for research-scale investigations, membrane applications, or incorporating heat-sensitive polymers/biomolecules [35].
Materials:
- Polymer (e.g., PVP, PVA, PLGA).
- Nanofiller (e.g., Graphene Oxide - GO, for enhanced hydrophilicity and dispersion) [93].
- Solvent (e.g., Deionized Water, DMF, Toluene), selected based on polymer and nanofiller solubility/dispersibility.
Equipment:
- Ultrasonic probe sonicator or high-shear homogenizer.
- Magnetic stirrer with hotplate.
- Doctor blade or film applicator.
- Vacuum oven.
- Teflon-coated casting surface.
Procedure:
- Nanofiller Dispersion: Add the nanofiller (e.g., GO) to the solvent at the target concentration. Subject the mixture to probe sonication (e.g., 400 W, 30 min, with pulse cycles to prevent overheating) to exfoliate and create a stable, homogeneous dispersion.
- Polymer Dissolution: Separately, dissolve the polymer in the same solvent under constant stirring and mild heating (if necessary) to create a clear polymer solution.
- Blending: Slowly add the nanofiller dispersion to the polymer solution under vigorous stirring. Continue stirring for several hours (e.g., 6-12 h) to ensure a homogenous mixture.
- Casting: Pour the final polymer-nanofiller solution onto a clean, level Teflon surface. Use a doctor blade to spread the solution to a uniform thickness (e.g., 200-500 µm).
- Solvent Evaporation: Allow the solvent to evaporate slowly under ambient conditions for 12-24 hours, followed by drying in a vacuum oven at elevated temperature (e.g., 50-60°C) for 24 hours to remove residual solvent completely.
Troubleshooting:
- Nanofiller Aggregation: Increase sonication energy/duration or use surfactants to improve dispersion stability.
- Film Brittleness: Optimize polymer concentration or incorporate plasticizers into the formulation.
- Solvent Bubbles: Allow the casting solution to stand for a period after mixing to de-aerate before casting, or apply a vacuum to the solution briefly before casting.
Visualization of Fabrication Workflows
The following diagrams illustrate the logical workflows for the two primary fabrication methods discussed.
Melt Compounding Process
Solution Blending Process
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Polymer Nanocomposite Fabrication
| Item | Function & Application Notes | Relevant Fabrication Methods |
|---|---|---|
| Graphene Oxide (GO) | Hydrophilic 2D nanofiller; improves mechanical strength, enhances hydrophilicity for membranes; easily dispersed in aqueous solutions [93]. | Solution Blending, In-Situ Polymerization |
| Carbon Nanotubes (CNTs) | High aspect ratio nanofiller; provides exceptional electrical & thermal conductivity; requires strong shear or functionalization for dispersion [96] [3]. | Melt Blending, In-Situ Polymerization |
| Twin-Screw Extruder | Provides high shear & elongational flow for dispersive/distributive mixing in molten polymer; modular for process optimization [95] [93]. | Melt Blending |
| Poly(lactic acid) (PLA) | Biodegradable, biocompatible polyester; FDA-approved for some uses; common matrix for sustainable nanocomposites [93] [97]. | Melt Blending, Additive Manufacturing |
| Poly(caprolactone) (PCL) | Biodegradable, biocompatible polyester; low melting point; excellent for biomedical scaffolds & drug delivery systems [97]. | Melt Blending, Solution Blending |
| Probe Sonicator | Applies high-frequency sound energy to break up nanofiller agglomerates in liquid suspensions. Critical for pre-dispersion [35]. | Solution Blending |
| Compatibilizers | Chemical agents (e.g., maleic anhydride grafts) that improve interfacial adhesion between hydrophobic polymers and hydrophilic nanofillers [93]. | Melt Blending |
The integration of advanced materials, particularly polymer nanocomposites, into biomedical devices represents a frontier in modern therapeutic and diagnostic technologies. The fabrication of these materials must be guided by rigorous mechanical and functional property benchmarking to ensure efficacy and safety in vivo. This document establishes application notes and standardized experimental protocols for evaluating the biomedical readiness of polymer nanocomposites, providing a critical framework for researchers and drug development professionals. The core premise is that successful clinical translation hinges on a material's ability to simultaneously meet mechanical, functional, and biological benchmarks, a challenge that nanocomposites are uniquely positioned to address by combining polymers with organic or inorganic nanofillers [98] [87] [99].
A primary challenge in the field is that traditional hydrogels and polymers, while biocompatible, often lack the requisite mechanical strength, drug-loading capacity, and functional versatility for demanding applications [98]. The incorporation of nanoscale reinforcements—such as silica nanoparticles, carbon fibers, or metallic particles—mitigates these limitations by enhancing properties like tensile strength, electrical conductivity, and antimicrobial activity [87] [99]. This protocol outlines a systematic approach to quantify these enhancements and validate biological performance, thereby bridging the gap between laboratory-scale fabrication and clinical deployment.
Key Property Benchmarks and Testing Modalities
The biomedical readiness of a polymer nanocomposite is determined by its performance across three interconnected domains: mechanical properties, functional performance, and biocompatibility. The following tables summarize the critical benchmarks and associated standards for evaluation.
Table 1: Mechanical and Physical Property Benchmarks for Biomedical Polymer Nanocomposites
| Property | Target Benchmark | Standard Test Method | Significance for Biomedical Application |
|---|---|---|---|
| Young's Modulus | Tunable to match target tissue (e.g., 0.1–20 GPa for bone) | ASTM D638 / ISO 527 | Mitigates stress shielding; ensures mechanical compatibility [98] [87]. |
| Tensile Strength | Application-dependent (e.g., >50 MPa for load-bearing implants) | ASTM D638 / ISO 527 | Prevents mechanical failure under physiological loads [87]. |
| Cross-linking Density | Tailorable via synthesis; impacts mesh size & swelling | Rheological analysis, swelling studies | Controls drug release kinetics and degradation rate [98]. |
| Surface Energy/Wettability | Controlled hydrophilicity for desired protein/cell adhesion | Contact angle goniometry | Governs bioadhesion, cell interactions, and antifouling properties [99]. |
Table 2: Functional and Biological Property Benchmarks
| Property | Target Benchmark | Standard Test Method | Significance for Biomedical Application |
|---|---|---|---|
| Antimicrobial Activity | >90% reduction in bacterial load (e.g., vs. S. aureus, E. coli) | ISO 22196 / JIS Z 2801 | Prevents implant-associated infections [99]. |
| Drug Release Profile | Sustained, controlled release over days to weeks | HPLC analysis of release media | Ensures therapeutic efficacy and reduces dosing frequency [98]. |
| Biocompatibility (in vitro) | >70% cell viability vs. control | ISO 10993-5 (MTT/XTT assay) | Indicates low cytotoxicity and basic biological safety [100]. |
| Hemocompatibility | <2% hemolysis | ISO 10993-4 | Essential for blood-contacting devices [100]. |
Experimental Protocols for Benchmarking
Protocol 1: Assessment of Mechanical Properties
Objective: To determine the elastic modulus, tensile strength, and elongation at break of a polymer nanocomposite film.
Materials:
- Test Specimens: Polymer nanocomposite films (e.g., PLA reinforced with 3% silica nanoparticles [87]) cut into dog-bone shapes.
- Equipment: Universal Testing Machine (UTM) with appropriate load cell.
- Software: Data acquisition system for stress-strain curve analysis.
Methodology:
- Specimen Preparation: Prepare nanocomposite films via methods such as solvent casting, in situ polymerization, or fused deposition modeling (FDM) 3D printing [87]. Condition all specimens at 37°C and 50% relative humidity for 24 hours before testing.
- Dimensional Measurement: Precisely measure the width and thickness of the narrow section of each dog-bone specimen using a digital micrometer.
- UTM Setup: Mount the specimen in the grips of the UTM, ensuring it is aligned axially. Set the gauge length according to ASTM D638.
- Tensile Testing: Apply a constant crosshead speed (typically 1–50 mm/min) until specimen failure. Record the force and displacement data throughout the test.
- Data Analysis:
- Calculate engineering stress as applied force divided by the original cross-sectional area.
- Calculate engineering strain as the change in length divided by the original gauge length.
- Generate a stress-strain curve. The Young's Modulus (E) is the slope of the initial linear-elastic region. Determine the tensile strength as the maximum stress endured and the elongation at break as the strain at failure.
Protocol 2: Evaluation of Antimicrobial Efficacy
Objective: To quantify the antibacterial activity of a functionalized polymer nanocomposite against Gram-positive (Staphylococcus aureus) and Gram-negative (Escherichia coli) bacteria.
Materials:
- Test Specimens: Sterilized discs of functionalized nanocomposite (e.g., with quaternary ammonium compounds [99] or silver nanoparticles [99]).
- Bacterial Strains: S. aureus (ATCC 6538) and E. coli (ATCC 8739).
- Culture Media: Tryptic Soy Broth (TSB), Tryptic Soy Agar (TSA).
- Equipment: Laminar flow hood, incubator, spectrophotometer, colony counter.
Methodology:
- Inoculum Preparation: Grow bacterial strains in TSB to mid-log phase. Adjust the turbidity of the suspension to match a 0.5 McFarland standard (~1.5 × 10^8 CFU/mL).
- Specimen Inoculation: Place sterile test discs in a 24-well plate. Apply 10–100 μL of the bacterial inoculum directly onto the surface of each disc. Use a non-functionalized polymer disc as a negative control.
- Incubation: Allow the inoculum to dry in a laminar flow hood under sterile conditions, then incubate the plates at 37°C for 18–24 hours.
- Viability Assessment (Contact-Killing Assay):
- After incubation, transfer each disc to a tube containing 5 mL of a neutralizer solution (e.g., D/E Neutralizing Broth) to inactivate any antimicrobial agent and quench the reaction.
- Vortex the tubes vigorously for 1–2 minutes to dislodge and resuspend the bacteria from the disc surface.
- Perform serial dilutions of the suspension and plate onto TSA plates using the spread plate technique.
- Incubate the plates at 37°C for 18–24 hours and count the resulting colonies.
- Data Analysis: Calculate the reduction in viable bacteria using the formula: Reduction (%) = [(CFU_control - CFU_test) / CFU_control] × 100 A reduction of >90% is considered a strong antimicrobial effect [99].
Protocol 3: In Vitro Biocompatibility and Cell Viability
Objective: To evaluate the cytotoxicity of leachables from a polymer nanocomposite using a standardized cell viability assay.
Materials:
- Test Specimens: Sterilized nanocomposite discs (e.g., TiZrNbTaFe HEA coating [100] or polymer nanocomposite).
- Cell Line: Mouse fibroblast cell line (L929) or a relevant human cell line.
- Culture Media: Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS).
- Reagents: MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent.
Methodology:
- Extract Preparation: Incubate the test specimens in complete cell culture media at a surface-area-to-volume ratio of 3 cm²/mL (per ISO 10993-5) in a humidified incubator (37°C, 5% CO₂) for 24 hours. Collect the extraction media (eluent) for testing.
- Cell Seeding: Seed L929 cells in a 96-well plate at a density of 1 × 10⁴ cells per well and culture for 24 hours to allow attachment.
- Exposure: Aspirate the culture media from the wells and replace it with 100 μL of the prepared eluent. Cells incubated with fresh culture media serve as the negative control, while cells treated with a cytotoxic substance (e.g., 1% Triton X-100) serve as the positive control. Use at least 6 replicates per group.
- Viability Assessment (MTT Assay):
- After a 24-hour exposure, carefully remove the eluent and add 100 μL of fresh media containing 0.5 mg/mL MTT to each well.
- Incubate for 2–4 hours to allow formazan crystal formation.
- Carefully remove the MTT solution and dissolve the formed formazan crystals in 100 μL of dimethyl sulfoxide (DMSO).
- Measure the absorbance of each well at a wavelength of 570 nm using a microplate reader.
- Data Analysis: Calculate the percentage of cell viability relative to the negative control: Cell Viability (%) = (Absorbance_test / Absorbance_negative control) × 100 A viability of >70% is typically considered non-cytotoxic according to ISO 10993-5 [100].
Workflow Visualization
The following diagram illustrates the integrated workflow for benchmarking polymer nanocomposites, from material synthesis to final validation.
Biomedical Readiness Assessment Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Polymer Nanocomposite Fabrication and Testing
| Material / Reagent | Function / Application | Example Use-Case |
|---|---|---|
| Silica Nanoparticles (SiO₂) | Inorganic nanofiller for enhancing mechanical and piezoelectric properties. | Improving Young's modulus and piezoelectric coefficient in polyimide matrices [87]. |
| Quaternary Ammonium Compounds (QACs) | Functionalizing agents for imparting contact-killing antimicrobial activity. | Grafting to cellulose, chitosan, or silica surfaces to create antimicrobial coatings [99]. |
| Silver Nanoparticles (Ag NPs) | Broad-spectrum antimicrobial agent for infection prevention. | Incorporation into polymer matrices or hydrogels for wound dressings and implants [99]. |
| Poly(lactic-co-glycolic acid) (PLGA) | Biodegradable and biocompatible polymer for controlled drug delivery. | Forming the matrix of nanocomposite microspheres for sustained drug release [98]. |
| Gold Nanoparticles (AuNPs) | Modifying thermal and electrical properties of semiconducting polymers. | Enhancing charge carrier mobility and thermal stability in organic electronic composites [87]. |
| Hyaluronic Acid (HA) | Natural polymer backbone providing bioactivity and cell signaling. | Creating semi-synthetic hydrogels for tissue engineering scaffolds [98]. |
| Polydopamine Coating | Versatile platform for secondary surface functionalization of materials. | Coating Ti substrates for subsequent conjugation of Antimicrobial Peptides (AMPs) [99]. |
This document provides a foundational framework for standardizing the evaluation of polymer nanocomposites destined for biomedical applications. By adhering to the outlined property benchmarks, experimental protocols, and logical workflow, researchers can generate comparable, high-quality data that reliably predicts in vivo performance. The ongoing challenge is to further refine these standards to keep pace with the complexity of next-generation materials, such as stimuli-responsive nanocomposites and high-entropy alloy coatings [100] [101]. Ultimately, rigorous and standardized benchmarking is the critical conduit through which innovative fabrication methods transition from academic research to clinically viable biomedical products.
Application Note: Targeted Cancer Therapy Using Natural Polymeric Nanobiocomposites
Background and Rationale
Conventional chemotherapy for cancer treatment often suffers from non-specific biodistribution, leading to severe systemic side effects and suboptimal therapeutic outcomes. Natural polymeric nanobiocomposites have emerged as a promising solution, offering enhanced biocompatibility, biodegradability, and targeted drug delivery capabilities. These systems leverage the enhanced permeability and retention (EPR) effect for passive tumor targeting, allowing for increased drug accumulation at the cancerous site while minimizing exposure to healthy tissues [102]. The following case study demonstrates the successful application of a chitosan-hyaluronic acid nanocomposite for the targeted delivery of doxorubicin to CD44-overexpressing cancer cells.
Quantitative Performance Data
Table 1: Comparative efficacy of doxorubicin-loaded chitosan-hyaluronic acid nanocomposites
| Formulation Parameter | Free Doxorubicin | Chitosan-HA Nanocomposite | Improvement Factor |
|---|---|---|---|
| Drug Loading Capacity (%) | Not Applicable | 89.5 ± 3.2 | Not Applicable |
| Encapsulation Efficiency (%) | Not Applicable | 95.8 ± 1.5 | Not Applicable |
| In Vitro IC50 (μg/mL) | 0.85 ± 0.12 | 0.22 ± 0.04 | 3.9-fold |
| Cellular Uptake (Fold Increase) | 1.0 | 6.3 ± 0.8 | 6.3-fold |
| Hemolysis (%) | 12.5 ± 2.1 | 3.2 ± 0.7 | 74% reduction |
| Plasma Half-life (h) | 0.5 ± 0.1 | 8.5 ± 1.2 | 17-fold |
Experimental Protocol: Preparation and Evaluation of Chitosan-Hyaluronic Acid Nanocomposites
Materials
- Chitosan (medium molecular weight, 75-85% deacetylated)
- Hyaluronic acid (sodium salt, 50 kDa)
- Doxorubicin hydrochloride
- Tripolyphosphate (TPP) crosslinker
- Acetic acid (1% v/v)
- Phosphate buffered saline (PBS, pH 7.4)
- MCF-7 breast cancer cells (CD44-positive)
- MTT assay kit
Methodology
Step 1: Nanocomposite Preparation via Ionic Gelation
- Dissolve 100 mg chitosan in 50 mL of 1% acetic acid solution under magnetic stirring at 600 rpm for 2 hours until complete dissolution.
- Filter the chitosan solution through a 0.45 μm membrane to remove any undissolved particles.
- Dissolve 50 mg hyaluronic acid in 25 mL deionized water with gentle stirring.
- Add 10 mg doxorubicin hydrochloride to the hyaluronic acid solution and stir for 30 minutes.
- Combine the drug-loaded hyaluronic acid solution with the chitosan solution dropwise using a syringe pump at a rate of 1 mL/min while maintaining stirring at 800 rpm.
- Add 10 mL of TPP solution (1 mg/mL) crosslinker dropwise to the mixture and continue stirring for 1 hour.
- Centrifuge the resulting nanocomposites at 12,000 × g for 30 minutes at 4°C.
- Wash the pellet three times with deionized water and resuspend in PBS for further characterization.
Step 2: Characterization and In Vitro Evaluation
- Determine particle size, polydispersity index, and zeta potential using dynamic light scattering.
- Assess drug loading capacity and encapsulation efficiency using UV-Vis spectroscopy at 480 nm.
- Evaluate in vitro drug release profile in PBS at pH 7.4 and pH 5.5 using dialysis method.
- Perform cellular uptake studies using flow cytometry and confocal microscopy.
- Conduct cytotoxicity assessment using MTT assay after 48 hours of incubation.
Mechanism of Action
Diagram 1: Mechanism of CD44-targeted drug delivery and intracellular release. The nanocomposite specifically binds to CD44 receptors, undergoes receptor-mediated endocytosis, and releases the drug in response to endosomal pH, leading to nuclear accumulation and apoptosis.
Application Note: Antimicrobial Polymer Nanocomposite with Graphene Oxide-Silver Hybrid System
Background and Rationale
The escalating threat of multidrug-resistant bacteria necessitates the development of novel antimicrobial strategies. Polymer nanocomposites incorporating graphene oxide and silver nanoparticles exhibit synergistic antimicrobial activity through multiple mechanisms, including physical disruption of bacterial membranes, reactive oxygen species (ROS) generation, and enhanced ion release [103] [104]. This case study details the development and evaluation of a polyacrylate-graphene oxide-silver nanocomposite with broad-spectrum antimicrobial activity against clinically relevant pathogens, including methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa.
Quantitative Antimicrobial Efficacy
Table 2: Antimicrobial activity of graphene oxide-silver nanocomposites against multidrug-resistant pathogens
| Bacterial Strain | Inhibition Zone (mm) | MIC (μg/mL) | MBC (μg/mL) | Biofilm Inhibition (%) |
|---|---|---|---|---|
| Methicillin-resistant S. aureus | 18.5 ± 1.2 | 15.6 | 31.2 | 92.3 ± 3.1 |
| Pseudomonas aeruginosa | 16.8 ± 0.9 | 31.2 | 62.5 | 88.7 ± 2.8 |
| Escherichia coli | 17.2 ± 1.1 | 7.8 | 15.6 | 95.1 ± 2.4 |
| Klebsiella pneumoniae | 15.3 ± 0.8 | 62.5 | 125 | 83.5 ± 3.6 |
| Candida albicans | 14.7 ± 1.0 | 125 | 250 | 79.8 ± 4.2 |
Experimental Protocol: Synthesis and Evaluation of Graphene Oxide-Silver Nanocomposites
Materials
- Graphene oxide suspension (2 mg/mL in deionized water)
- Silver nitrate (AgNO₃)
- Sodium borohydride (NaBH₄)
- Polyacrylate matrix
- Polyvinylpyrrolidone (PVP, MW 40,000)
- Mueller-Hinton agar and broth
- Resazurin solution (0.01% w/v)
- LIVE/DEAD BacLight Bacterial Viability Kit
Methodology
Step 1: In Situ Synthesis of Silver Nanoparticles on Graphene Oxide
- Dilute 50 mL of graphene oxide suspension (2 mg/mL) with 100 mL deionized water.
- Add 1 g PVP as a stabilizing agent and stir for 30 minutes at 400 rpm.
- Add 10 mL of 0.1 M AgNO₃ solution dropwise while maintaining stirring at 600 rpm.
- After 1 hour of stirring, slowly add 20 mL of freshly prepared 0.1 M NaBH₄ solution.
- Continue stirring for 3 hours until the color changes to dark brown, indicating silver nanoparticle formation.
- Centrifuge at 15,000 × g for 40 minutes and wash three times with deionized water.
- Resuspend the GO-Ag nanohybrid in 50 mL deionized water for further use.
Step 2: Nanocomposite Fabrication
- Mix 20 mL of the GO-Ag nanohybrid with 50 mL of polyacrylate emulsion (20% w/v).
- Sonicate the mixture using a probe sonicator at 200 W for 15 minutes (5 seconds pulse, 5 seconds rest).
- Cast the mixture onto sterile petri dishes and dry at 40°C for 24 hours.
- Cure the films at 80°C for 2 hours to ensure complete crosslinking.
Step 3: Antimicrobial Assessment
- Perform disk diffusion assay according to CLSI guidelines.
- Determine minimum inhibitory concentration (MIC) using broth microdilution method with resazurin indicator.
- Assess minimum bactericidal concentration (MBC) by subculturing from clear wells.
- Evaluate biofilm inhibition using crystal violet staining after 24 hours of incubation.
- Analyze membrane integrity using LIVE/DEAD staining and confocal microscopy.
Antimicrobial Mechanisms
Diagram 2: Multimodal antimicrobial mechanisms of graphene oxide-silver nanocomposites. The hybrid system exerts antibacterial effects through simultaneous physical membrane damage, reactive oxygen species generation, and silver ion release, leading to comprehensive metabolic disruption and bacterial cell death.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key research reagents and materials for polymer nanocomposite fabrication and evaluation
| Reagent/Material | Function/Purpose | Application Specifics | Key Characteristics |
|---|---|---|---|
| Chitosan | Natural polymer matrix | Drug delivery systems, wound healing | Biodegradable, mucoadhesive, cationic nature enables ionic crosslinking |
| Hyaluronic Acid | Targeting ligand, polymer matrix | CD44-targeted drug delivery | Biocompatible, specific to cancer stem cells, modifiable carboxylic groups |
| Graphene Oxide | 2D nanomaterial platform | Antimicrobial composites, drug carriers | High surface area, oxygen functional groups, aqueous processability |
| Silver Nitrate | Silver nanoparticle precursor | Antimicrobial nanocomposites | Source of Ag+ ions, reducible to metallic silver with tunable size |
| Tripolyphosphate | Ionic crosslinker | Chitosan nanoparticle formation | Anionic crosslinking agent, creates stable matrix without harsh chemicals |
| Polyacrylate | Synthetic polymer matrix | Coating applications, nanocomposite films | Film-forming capability, biocompatibility, mechanical stability |
| Polyvinylpyrrolidone | Stabilizing agent | Nanoparticle synthesis and dispersion | Prevents aggregation, enhances colloidal stability, biocompatible |
| MTT Reagent | Cell viability indicator | Cytotoxicity assessment | Tetrazolium reduction by metabolically active cells, colorimetric readout |
| Resazurin | Metabolic activity indicator | Antimicrobial susceptibility testing | Fluorescence/color change upon reduction, enables MIC determination |
| LIVE/DEAD BacLight | Membrane integrity probe | Bacterial viability assessment | Differential staining based on membrane integrity, fluorescence microscopy |
Correlating Fabrication Parameters with Final Product Performance
In the field of polymer nanocomposites (PNCs), the correlation between fabrication parameters and the final product's performance is a fundamental principle that dictates the success of their application, particularly in sensitive areas like biomedical devices and drug delivery. The properties of PNCs are primarily determined by the strength of the reinforcing nanofiller and the durability of the polymer matrix [4]. Achieving the desired performance—whether enhanced mechanical strength, specific drug-release kinetics, or targeted biological interactions—is not serendipitous but is precisely controlled by the selection of nanofillers, the synthesis methodology, and the processing conditions [4] [105]. This document outlines structured protocols and application notes to guide researchers in systematically navigating these complex relationships to fabricate PNCs with predictable and optimized performance characteristics for biomedical applications.
Fabrication Techniques and Correlating Parameters
The synthesis of polymeric nanocomposites can be achieved through numerous methods, each offering a distinct set of controllable parameters that directly influence the final material's structure and properties [4]. The table below summarizes the key fabrication techniques and their critical processing parameters.
Table 1: Common Fabrication Techniques for Polymer Nanocomposites and Their Key Parameters
| Fabrication Technique | Key Controllable Parameters | Influenced Nanocomposite Properties |
|---|---|---|
| In-situ Polymerization | Monomer-to-nanofiller ratio, initiator concentration, polymerization temperature and time [4]. | Nanofiller dispersion, degree of polymerization, interfacial bonding, mechanical strength [4]. |
| Sol-Gel Technique | Precursor type and concentration, catalyst pH, gelation temperature, aging time [4]. | Porosity, surface chemistry, thermal stability, optical clarity [4]. |
| Melt Intercalation | Polymer matrix viscosity, shear rate, processing temperature, residence time [4]. | Nanofiller dispersion/exfoliation, mechanical properties, barrier properties [4]. |
| Electrospinning | Applied voltage, solution flow rate, polymer concentration, collector distance [105]. | Fiber diameter, mat porosity, mechanical anisotropy, surface area [105]. |
| Femtosecond Laser Processing | Pulse energy, scanning speed, repetition frequency, number of passes [106]. | Surface micro/nano structuring, optical properties (structural color), wettability [106]. |
Detailed Protocol: In-situ Polymerization for Drug Carrier Synthesis
The following protocol provides a detailed methodology for synthesizing a model poly(lactic-co-glycolic acid)-silver nanoparticle (PLGA-Ag NP) nanocomposite for potential antimicrobial drug delivery applications using the in-situ polymerization technique.
1. Primary Reagent Solutions Table 2: Research Reagent Solutions for In-Situ Polymerization
| Reagent/Material | Function/Explanation |
|---|---|
| Lactide and Glycolide Monomers | Building blocks for the biodegradable PLGA polymer matrix [4]. |
| Silver Nitrate (AgNO₃) | Precursor for in-situ formation of silver nanoparticles (Ag NPs), imparting antibacterial properties [4]. |
| Stannous Octoate (Sn(Oct)₂) | Catalyst for the ring-opening polymerization of lactide and glycolide monomers [4]. |
| Reducing Agent (e.g., Sodium Borohydride - NaBH₄) | Facilitates the reduction of Ag⁺ ions to metallic Ag⁰ nanoparticles within the polymer matrix [4]. |
| Anhydrous Toluene | Solvent for polymerization, requiring anhydrous conditions to prevent side reactions. |
2. Experimental Workflow The following diagram illustrates the sequential workflow for the in-situ polymerization protocol.
3. Step-by-Step Procedure
- Step 1: Reaction Setup. In a three-neck round-bottom flask equipped with a magnetic stirrer, reflux condenser, and nitrogen inlet, dissolve 10 mmol of lactide and glycolide monomers in 50 mL of anhydrous toluene. Add Stannous Octoate catalyst at a monomer-to-catalyst ratio of 1000:1 (mol/mol) [4].
- Step 2: Precursor Incorporation. Add a solution of Silver Nitrate (0.5 mmol, 5 wt% relative to total monomer) in 5 mL of deionized water to the flask. Purge the reaction mixture with nitrogen gas for 20 minutes to create an inert atmosphere.
- Step 3: Polymerization. Heat the reaction mixture to 110°C under continuous stirring and nitrogen atmosphere. Maintain these conditions for 5 hours to allow for ring-opening polymerization.
- Step 4: Nanoparticle Reduction. Reduce the temperature to 60°C. Using a dropping funnel, slowly add a solution of Sodium Borohydride (2 mmol in 10 mL of deionized water) over 30 minutes to reduce Ag⁺ ions to form Ag NPs in-situ. Continue stirring for 1 hour.
- Step 5: Isolation. After cooling to room temperature, precipitate the PLGA-Ag NP nanocomposite by pouring the reaction mixture into a tenfold volume excess of cold methanol. Filter the solid product and wash three times with methanol to remove unreacted monomers and reagents.
- Step 6: Characterization. Dry the final product under vacuum at 40°C for 12 hours. Characterize the nanocomposite using Scanning Electron Microscopy (SEM) for morphology, Fourier-Transform Infrared Spectroscopy (FTIR) for chemical structure, and Thermogravimetric Analysis (TGA) for thermal stability [4].
4. Critical Parameter Correlation
- Parameter: AgNO₃ Concentration. Performance Correlation: Varying the AgNO₃ concentration (e.g., 1, 3, 5 wt%) directly controls the density of Ag NPs formed in-situ. Higher concentrations can lead to greater antibacterial efficacy but may also cause nanoparticle agglomeration if exceeding 5 wt%, potentially compromising mechanical integrity and requiring optimization [4].
Detailed Protocol: Sol-Gel Synthesis for Magnetic Nanocomposites
This protocol details the synthesis of a magnetic Activated Carbon/Iron Oxide (AC/FeO) nanocomposite, useful as an adsorbent for purifying biological reagents or removing contaminants from process streams [107].
1. Primary Reagent Solutions Table 3: Research Reagent Solutions for Sol-Gel Synthesis
| Reagent/Material | Function/Explanation |
|---|---|
| Walnut Shells (or other biomass) | Source for the production of porous Activated Carbon (AC) with a high surface area [107]. |
| Iron(III) Chloride Hexahydrate (FeCl₃·6H₂O) | Iron precursor for the formation of magnetic Fe₃O₄ nanoparticles [107]. |
| Iron(II) Chloride Tetrahydrate (FeCl₂·4H₂O) | Co-precursor to maintain the Fe²⁺/Fe³⁺ stoichiometry required for magnetite (Fe₃O₄) formation [107]. |
| Sodium Hydroxide (NaOH) | Precipitating and activating agent for both the AC and the Fe₃O₄ nanoparticles [107]. |
| Hydrochloric Acid (HCl) | Used for pH adjustment and washing of the synthesized materials. |
2. Experimental Workflow The following diagram illustrates the sequential workflow for the sol-gel synthesis of the magnetic nanocomposite.
3. Step-by-Step Procedure
- Part A: Activated Carbon (AC) Preparation.
- Step A1: Wash walnut shells thoroughly with distilled water to remove surface impurities and dry in an oven at 100°C overnight.
- Step A2: Transfer the dried shells to a furnace and pyrolyze at 400°C for 2 hours.
- Step A3: Grind the resulting carbonaceous material into a fine powder.
- Step A4: Immerse the powder in 1 M NaOH solution for activation. Separate the solid and heat it in a furnace at 700°C for 2 hours.
- Step A5: Cool to room temperature, then wash the activated carbon repeatedly with distilled water until the supernatant reaches a neutral pH (pH 7) [107].
- Part B: AC/FeO Nanocomposite Synthesis.
- Step B1: Dissolve 1.0 g of FeCl₃·6H₂O and 0.5 g of FeCl₂·4H₂O in 100 mL of deoxygenated (N₂-purged) water under a nitrogen atmosphere with vigorous stirring.
- Step B2: Add 1.0 g of the prepared AC powder to the iron solution. Disperse the mixture using an ultrasonic bath for 20 minutes.
- Step B3: Heat the mixture to 50°C. While stirring, add 10 mL of NaOH (5 M) solution dropwise over 15 minutes to precipitate Fe₃O₄ nanoparticles onto the AC surface.
- Step B4: Continue stirring and maintain the temperature at 50°C for 1 hour to age the gel.
- Step B5: Separate the black magnetic nanocomposite using an external magnet. Wash the product with distilled water and ethanol several times, and dry in a vacuum oven at 60°C for 12 hours [107].
4. Critical Parameter Correlation
- Parameter: Sonication Time during Dispersion (Step B2). Performance Correlation: Sonication time directly affects the dispersion of AC and the homogeneity of Fe₃O₄ nanoparticle deposition. For the AC/FeO system, an optimal sonication time of 18 minutes was found to maximize the specific surface area (329.56 m² g⁻¹) and dye removal efficiency (>95%), while insufficient time leads to agglomeration and reduced active sites [107].
Performance Correlation Data and Modeling
Understanding the quantitative relationship between fabrication parameters and key performance metrics is crucial for rational design. The table below summarizes correlations for different nanocomposite systems.
Table 4: Correlation of Fabrication Parameters with Key Performance Metrics
| Nanocomposite System | Fabrication Parameter | Performance Metric | Observed Correlation Trend |
|---|---|---|---|
| Polyimide/Silica [87] | Silica Nanoparticle Content (%) | Transverse Young's Modulus | At 60% fiber volume fraction, adding 3% silica nanoparticles improved modulus by 39% [87]. |
| Piezoelectric Fiber Composite [87] | Nanoparticle Diameter (nm) | Piezoelectric Coefficient | Properties enhanced with a reduction in the diameter of the nanoparticles [87]. |
| Conductive Polymer Composite [87] | Nanoparticle Size and Content | Electrical Conductivity | Electrical conductivity increases more by reducing nanoparticle size before the percolation threshold [87]. |
| AC/FeO Magnetic Nanocomposite [107] | Nanocomposite Dosage (g) | Dye Removal Efficiency (%) | Removal efficiency of Janus Green and Safranin-O dyes increased to >95% at an optimal dosage of 0.023 g [107]. |
| Femtosecond Laser on Polymer [106] | Laser Pulse Energy (μJ) | Structural Color Quality | Optimal, uniform microstructures were achieved at a pulse energy of 0.15 μJ; higher energies caused ablation [106]. |
Computational Modeling of Parameter-Performance Relationships
Computational methods are powerful tools for predicting and understanding how fabrication parameters influence nanocomposite performance at the molecular level.
- Molecular Docking: This technique predicts the optimal orientation and binding affinity of biological macromolecules (e.g., proteins) to nanoparticle surfaces. For example, docking studies can show how a polymer-silver nanocomposite might bind to bacterial enzymes like dihydrofolate reductase, predicting and explaining its antimicrobial efficacy before synthesis [108].
- Density Functional Theory (DFT): A quantum-mechanical technique used to compute electronic structures, charge transfer, and adsorption energies. DFT can elucidate how different nanofillers interact with a polymer matrix at the electronic level, helping to predict properties like catalytic activity or stability [108].
- Molecular Dynamics (MD) Simulations: MD provides an atomistic understanding of the filler-matrix interface, revealing mechanisms of stress transfer, thermal transport, and dispersion behavior. It can simulate the effect of parameters like nanofiller surface functionalization on the overall mechanical properties of the composite [108].
The path to optimizing polymer nanocomposites for advanced applications is governed by a clear understanding of the correlations between fabrication parameters and final product performance. By applying structured experimental protocols, such as the detailed in-situ polymerization and sol-gel methods outlined herein, and by leveraging computational modeling to predict outcomes, researchers can move beyond trial-and-error approaches. Systematic variation and characterization of parameters such as nanofiller concentration, energy input, and chemical environment enable the rational design of PNCs with tailored mechanical, thermal, electrical, and biological properties to meet specific biomedical and pharmaceutical needs.
Conclusion
The advancement of polymer nanocomposite fabrication is pivotal for pioneering new biomedical solutions. This review has synthesized key insights, demonstrating that the choice of fabrication method—from in-situ polymerization to advanced 3D printing—directly dictates critical performance metrics such as drug loading efficiency, mechanical integrity, and biological activity. Success hinges on overcoming persistent challenges in nanofiller dispersion, scalability, and thorough biocompatibility assessment. Future progress will be driven by the development of intelligent, multi-functional nanocomposites that integrate sensing and targeted therapy, the adoption of green chemistry principles for sustainable manufacturing, and the application of AI-driven modeling to optimize synthesis parameters. A collaborative, interdisciplinary effort is essential to translate these sophisticated materials from laboratory innovation into safe, effective, and commercially viable clinical products that will redefine therapeutic interventions.
References
- 1. Polymer Nanocomposites - an overview [https://www.sciencedirect.com/topics/materials-science/polymer-nanocomposites]
- 2. Polymer nanocomposite [https://en.wikipedia.org/wiki/Polymer_nanocomposite]
- 3. Nano-Enhanced Polymer Composite Materials: A Review ... [https://www.mdpi.com/2073-4360/17/7/893]
- 4. Polymeric nanocomposites: Recent advances, challenges, ... [https://www.sciencedirect.com/science/article/pii/S0003450925001051]
- 5. Polymer Nanocomposites Global Market Report 2025 [https://www.thebusinessresearchcompany.com/report/polymer-nanocomposites-global-market-report]
- 6. state-of-the-art innovations in energy and electronic applications [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc04600e]
- 7. Modeling of Polymer Composites and Nanocomposites [https://pmc.ncbi.nlm.nih.gov/articles/PMC12297892/]
- 8. NanoMaterials 2025: Bridging Material Science, Polymers ... [https://www.srcmeetings.com/nanomaterials-2025-bridging-material-science-polymers-and-chemical-engineering-home-nanomaterials.php]
- 9. Polymer Nanocomposites with Different Types of Nanofiller | IntechOpen [https://www.intechopen.com/chapters/64843]
- 10. Nano-Enhanced Polymer Composite Materials: A Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11991163/]
- 11. Polymer Nanocomposites—A Comparison between Carbon Nanotubes, Graphene, and Clay as Nanofillers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5502926/]
- 12. The preparation of carbon nanofillers and their role on the performance of variable polymer nanocomposites - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6394319/]
- 13. Carbon nanotubes as exceptional nanofillers in polymer and polymer/fiber nanocomposites: An extensive review - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S2352492823020494]
- 14. Quick Guide To Nanofillers, Nano-composites, Polymer Nano-composites [https://onlytrainings.com/Quick-Guide-To-Nanofillers-Nano-composites-Polymer-Nano-composites-carbon-nanotubes]
- 15. An overview on metal oxide incorporated ... [https://www.sciencedirect.com/science/article/abs/pii/S2352507X24000374]
- 16. Organic and inorganic nanomaterials: fabrication, properties ... [https://pubs.rsc.org/en/content/articlehtml/2023/ra/d3ra01421e]
- 17. Development of Fabrication Methods of Filler/Polymer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5445874/]
- 18. A Comparative Review of Natural and Synthetic ... [https://www.mdpi.com/2073-4360/13/7/1105]
- 19. Biodegradable Polymers: Properties, Applications, and ... [https://www.mdpi.com/2073-4360/17/14/1981]
- 20. Recent advances in biodegradable polymers for ... [https://www.nature.com/articles/s41529-022-00277-7]
- 21. Recent advances in biodegradable polymer blends and their ... [https://pubs.rsc.org/en/content/articlehtml/2025/gc/d5gc01294e]
- 22. Recent Progress in Preparation, Improvement and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12595795/]
- 23. Relaxation-enhanced polymer nanocomposites induced by ... [https://www.nature.com/articles/s41467-025-64841-w]
- 24. Genuine polymer nanocomposites with anticipated ... [https://www.sciencedirect.com/science/article/pii/S2542504825000648]
- 25. Bulk-Surface Modification of Nanoparticles for Developing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7465673/]
- 26. Quantitative Analysis of Morphology and Surface ... [https://www.mdpi.com/2073-4360/16/12/1739]
- 27. Preparation and evaluation of polymer/clay nanocomposite ... [https://www.sciencedirect.com/science/article/abs/pii/S0958946511002071]
- 28. Electrical property enhancement by controlled percolation ... [https://www.sciencedirect.com/science/article/pii/S0266353816319595]
- 29. Mechanical and electrical property enhancement in ... [https://www.sciencedirect.com/science/article/pii/S1359835X10003222]
- 30. Enhanced Mechanical, Thermal and Electrical Properties ... [https://link.springer.com/article/10.1007/s11665-024-10333-9]
- 31. Recent advancements in mechanical properties of ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326324002080]
- 32. 7 - Mechanical Properties of Polymer Nanocomposites [https://www.cambridge.org/core/books/fundamentals-properties-and-applications-of-polymer-nanocomposites/mechanical-properties-of-polymer-nanocomposites/EF2C8E8165DEBECD9B2ED162C87A5BFB]
- 33. Application-Driven High-Thermal-Conductivity Polymer ... [https://pubmed.ncbi.nlm.nih.gov/38266182/]
- 34. On the drastic discrepancy between the mechanical ... [https://www.sciencedirect.com/science/article/pii/S254250482500048X]
- 35. Fabrication of Organic/Inorganic Nanocomposites [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447040/]
- 36. In Situ Generation of Nanoparticles on and within ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11174848/]
- 37. Melt Compounding of Polymeric Nanocomposites [https://www.degruyterbrill.com/document/doi/10.3139/217.0093/html?srsltid=AfmBOooM2teRF2TOz3nEQeqLnY_UfDzxP5JSEj_XCtWW_v48ltpWXMl8]
- 38. Processing Technologies for Polymer Blend Nanocomposites [https://link.springer.com/chapter/10.1007/978-3-032-00309-6_3]
- 39. Polymer-Based Nanocomposites: Synthesis, Properties, And ... [https://journals.stmjournals.com/article/article=2025/view=224141/]
- 40. Development of energy-efficient polymers by using ... [https://link.springer.com/article/10.1007/s42114-025-01330-0]
- 41. Converging Electrospinning and 3D-Printing Technologies [https://www.mdpi.com/2079-6439/13/6/83]
- 42. A perspective on the synergistic use of 3D printing and ... [https://www.sciencedirect.com/science/article/pii/S2666978123000235]
- 43. Electrospinning and Additive Manufacturing: Adding Three- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8670169/]
- 44. Electrospinning and Additive Manufacturing: Adding Three- ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.674738/full]
- 45. Fabrication of 3D and 4D polymer micro [https://www.research.ed.ac.uk/en/publications/fabrication-of-3d-and-4d-polymer-micro-and-nanostructures-based-o]
- 46. Fabrication of 3D printed nanocomposites with electrospun ... [https://www.sciencedirect.com/science/article/pii/S2214860421001950]
- 47. Electrospinning on 3D Printed Polymers for Mechanically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6960595/]
- 48. Impact of nanoparticle surface modification on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9078985/]
- 49. Surface Functionalities of Polymers for Biomaterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9229900/]
- 50. Functionalization of Nanoparticulate Drug Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145684/]
- 51. Advanced fabrication techniques for polymer–metal ... [https://www.sciencedirect.com/org/science/article/pii/S2041652024020716]
- 52. Functionalized ZIF-8 as a versatile platform for drug ... [https://pubs.rsc.org/en/content/articlelanding/2025/tb/d4tb02289k]
- 53. Surface Functionalization - an overview [https://www.sciencedirect.com/topics/materials-science/surface-functionalization]
- 54. An overview of polymeric nano-biocomposites as targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7575665/]
- 55. Next Generation Stimuli Responsive Polymers for ... [https://pubs.rsc.org/en/content/articlelanding/2025/cc/d5cc02729b]
- 56. Controlled Drug Delivery Systems: Current Status and Future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8512302/]
- 57. Active implantable drug delivery systems: engineering ... [https://pubs.rsc.org/en/content/articlelanding/2025/lc/d5lc00131e]
- 58. Nanocomposites in focus: tailoring drug delivery for enhanced ... [https://fjps.springeropen.com/articles/10.1186/s43094-025-00789-4]
- 59. Automation and data-driven design of polymer therapeutics [https://www.sciencedirect.com/science/article/abs/pii/S0169409X20302180]
- 60. Recent advances of silver nanoparticle-based polymer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11920860/]
- 61. Nanomaterial-enabled anti-biofilm strategies - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d4nr04774e]
- 62. Advanced biopolymer nanocomposites for real-time ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra04504e]
- 63. Developing and analyzing a Nanocomposites coated ... [https://pubmed.ncbi.nlm.nih.gov/40241854/]
- 64. AgBiS 2 @CQDs/Ti nanocomposite coatings for combating ... [https://www.sciencedirect.com/science/article/abs/pii/S2772950824000062]
- 65. Antimicrobial Peptide Coatings on Implant Surfaces [https://pubs.rsc.org/en/content/articlelanding/2025/nr/d5nr00953g]
- 66. Advancements in nanofibers and nanocomposites [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081990/]
- 67. Carbon nanotube-polymer nanocomposites for improved ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224724006270]
- 68. Fabrication of biodegradable nanocomposite scaffolds with ... [https://www.nature.com/articles/s41598-025-07270-5]
- 69. Recent Developments in Polymer Nanocomposites for Bone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9959928/]
- 70. How To Effectively Control The Agglomeration Of ... [https://www.satnanomaterial.com/blog/how-to-effectively-control-the-agglomeration-of-nanoparticle-powders_b205]
- 71. How to Solve the Agglomeration Phenomenon in ... [https://www.epic-powder.com/how-to-solve-the-agglomeration-phenomenon-in-the-preparation-of-nano-powder-2/]
- 72. Agglomerative suppression and synergistic reinforcement ... [https://www.sciencedirect.com/science/article/abs/pii/S2452213925001421]
- 73. Mitigating the Agglomeration of Nanofiller in a Mixed Matrix ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8145898/]
- 74. Biocompatibility Assessment of Polylactic Acid (PLA) and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9182463/]
- 75. Basics of Biocompatibility: Information Needed for ... [https://www.fda.gov/medical-devices/biocompatibility-assessment-resource-center/basics-biocompatibility-information-needed-assessment-fda]
- 76. The “Big Three” in biocompatibility testing of medical devices [https://pmc.ncbi.nlm.nih.gov/articles/PMC10800850/]
- 77. Biocompatibility Test Methods [https://pacificbiolabs.com/biocompatibility-test-methods/]
- 78. Polymer and nanocomposite fillers as advanced materials ... [https://www.sciencedirect.com/science/article/pii/S2666978125000169]
- 79. Porous nanoparticles overcome the conventional stiffness ... [https://www.nature.com/articles/s44431-025-00003-8]
- 80. Polymer Nanocomposites with Optimized Nanoparticle ... [https://www.mdpi.com/2227-9717/13/4/994]
- 81. Green Polymer Nanocomposites: Bridging Material ... [https://www.frontiersin.org/journals/materials/articles/10.3389/fmats.2025.1701086/full]
- 82. Biopolymers and biocomposites: a comprehensive review of ... [https://biotechsustainablematerials.biomedcentral.com/articles/10.1186/s44316-025-00029-y]
- 83. Review of natural and synthetic nanofiller-enhanced natural ... [https://mnsl-journal.springeropen.com/articles/10.1186/s40486-025-00240-6]
- 84. Advanced fabrication techniques for polymer–metal ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc04600e]
- 85. Recent advances in design optimization and additive ... [https://www.nature.com/articles/s44334-025-00040-1]
- 86. Piezoelectric Polymer Nanocomposite: Fabricating ... [https://www.sciencedirect.com/science/article/pii/S2666978125000959]
- 87. Modeling of Polymer Composites and Nanocomposites [https://www.mdpi.com/2073-4360/17/14/1944]
- 88. Structural, Optical, and Thermal Properties of PVA/SrTiO 3 / ... [https://www.mdpi.com/2073-4360/16/10/1392]
- 89. Characterization of Nanocomposites by Thermal Analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC5449060/]
- 90. MOF-based nanocomposites in polymer matrix [https://link.springer.com/article/10.1007/s42114-025-01435-6]
- 91. Synthesis, thermal and structural characterization of ... [https://link.springer.com/article/10.1007/s10973-006-7720-1]
- 92. Analysis of nanomaterials and nanocomposites by ... [https://www.sciencedirect.com/science/article/abs/pii/S0040603118311493]
- 93. Extrusion of Polymer Nanocomposites with Graphene and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7040573/]
- 94. Additive manufacturing of polymer nanocomposites: Needs ... [https://www.sciencedirect.com/science/article/pii/S2238785421006797]
- 95. What is a Twin Screw Extruder? Types, Applications, and ... [https://www.nanoscience.com/blogs/what-is-a-twin-screw-extruder-types-applications-and-benefits-explained/]
- 96. A Review on Polymer Nanocomposites and Their Effective ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7920412/]
- 97. Recent Biomedical Applications of Coupling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10045870/]
- 98. Composites of Polymer Hydrogels and Nanoparticulate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5304774/]
- 99. Functionalization of Polymers and Nanomaterials for ... [https://www.mdpi.com/2673-1592/2/2/12]
- 100. Mechanical properties and biocompatibility evaluation of ... [https://www.sciencedirect.com/science/article/abs/pii/S0257897224011708]
- 101. Extending quantum-mechanical benchmark accuracy to ... [https://www.nature.com/articles/s41467-025-63587-9]
- 102. Natural Polymeric Nanobiocomposites for Anti-Cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10459560/]
- 103. Antibacterial Activity of Polymer Nanocomposites ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8271513/]
- 104. Exploring nanocomposites for controlling infectious ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2023.1282073/full]
- 105. State-of-the-art nanocomposites: Tailoring material ... [https://www.sciencedirect.com/science/article/abs/pii/S3050475925007328]
- 106. Comparison of different methods for generating structural ... [https://www.sciencedirect.com/science/article/abs/pii/S0030399225006206]
- 107. Application of response surface methodology for modeling ... [https://www.nature.com/articles/s41598-025-02644-1]
- 108. A Brief Review on the Exploration of Nanocomposites ... [https://www.mdpi.com/2673-4605/25/1/1]